
Mit snapgene Crack Key For U - apologise, can
Computational approaches for effective CRISPR guide RNA design and evaluation
Guanqing Liu,a,bYong Zhang,a,c,⁎ and Tao Zhanga,b,d,⁎
Guanqing Liu
aJiangsu Key Laboratory of Crop Genetics and Physiology/ Key Laboratory of Plant Functional Genomics of the Ministry of Education/ Jiangsu Key Laboratory of Crop Genomics and Molecular Breeding, Agricultural College of Yangzhou University, Yangzhou 225009, China
bJiangsu Co-Innovation Center for Modern Production Technology of Grain Crops, Yangzhou University, Yangzhou 225009, China
Find articles by Guanqing Liu
Yong Zhang
aJiangsu Key Laboratory of Crop Genetics and Physiology/ Key Laboratory of Plant Functional Genomics of the Ministry of Education/ Jiangsu Key Laboratory of Crop Genomics and Molecular Breeding, Agricultural College of Yangzhou University, Yangzhou 225009, China
cDepartment of Biotechnology, School of Life Sciences and Technology, Center for Informational Biology, University of Electronic Science and Technology of China, Chengdu 610054, China
Find articles by Yong Zhang
Tao Zhang
aJiangsu Key Laboratory of Crop Genetics and Physiology/ Key Laboratory of Plant Functional Genomics of the Ministry of Education/ Jiangsu Key Laboratory of Crop Genomics and Molecular Breeding, Agricultural College of Yangzhou University, Yangzhou 225009, China
bJiangsu Co-Innovation Center for Modern Production Technology of Grain Crops, Yangzhou University, Yangzhou 225009, China
dJoint International Research Laboratory of Agriculture and Agri-Product Safety, The Ministry of Education of China, Yangzhou University, Yangzhou 225009, China
Find articles by Tao Zhang
Author informationArticle notesCopyright and License informationDisclaimer
aJiangsu Key Laboratory of Crop Genetics and Physiology/ Key Laboratory of Plant Functional Genomics of the Ministry of Education/ Jiangsu Key Laboratory of Crop Genomics and Molecular Breeding, Agricultural College of Yangzhou University, Yangzhou 225009, China
bJiangsu Co-Innovation Center for Modern Production Technology of Grain Crops, Yangzhou University, Yangzhou 225009, China
cDepartment of Biotechnology, School of Life Sciences and Technology, Center for Informational Biology, University of Electronic Science and Technology of China, Chengdu 610054, China
dJoint International Research Laboratory of Agriculture and Agri-Product Safety, The Ministry of Education of China, Yangzhou University, Yangzhou 225009, China
Yong Zhang: nc.ude.ctseu@619gnoygnahz; Tao Zhang: nc.ude.uzy@oatgnahz
⁎Corresponding authors. nc.ude.ctseu@619gnoygnahz, nc.ude.uzy@oatgnahz
Received 2019 Aug 29; Revised 2019 Nov 9; Accepted 2019 Nov 15.
Copyright © 2019 The Authors
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Abstract
The Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR)/ CRISPR-associated (Cas) system has emerged as the main technology for gene editing. Successful editing by CRISPR requires an appropriate Cas protein and guide RNA. However, low cleavage efficiency and off-target effects hamper the development and application of CRISPR/Cas systems. To predict cleavage efficiency and specificity, numerous computational approaches have been developed for scoring guide RNAs. Most scores are empirical or trained by experimental datasets, and scores are implemented using various computational methods. Herein, we discuss these approaches, focusing mainly on the features or computational methods they utilise. Furthermore, we summarise these tools and give some suggestions for their usage. We also recommend three versatile web-based tools with user-friendly interfaces and preferable functions. The review provides a comprehensive and up-to-date overview of computational approaches for guide RNA design that could help users to select the optimal tools for their research.
1. Introduction
Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR)/CRISPR-associated (Cas) systems, such as Cas9 [46] and Cas12a (formerly Cpf1) [118], are the primary tools used for genome editing due to their various abilities to manipulate, detect, and image certain DNA and RNA sequences in the cell [50]. The CRISPR/Cas system was first adapted for genome editing in 2012 [31], [46], and subsequent studies have transformed the CRISPR RNA (crRNA) and trans-activating crRNA (tracrRNA) into a single guide RNA (sgRNA) that can bind to both the Cas9 protein and the target DNA sequence. Cas9 protein and sgRNA complex first scans the appropriate PAM sequence and binds to the targeted genome loci, then the activated HNH and the RuvC nuclease domain of Cas9 function to make a DNA double-strand break (DSB) in the specific region [22], [69].
The most frequently used CRISPR nuclease is type II endonuclease Cas9, which recognises the 5′-NGG-3′ PAM (SpCas9) [73]. Another popular nuclease is CRISPR type V endonuclease Cas12a (Cpf1), which recognises the 5′-TTTV-3′ PAM, and shows high efficiency in both animal and plant organisms [25], [48], [49], [71], [97], [118], [124]. Recently, several other Cas family proteins have also been discovered and adapted for DNA or RNA editing events, including Cas12b, Cas13a and Cas14 [2], [37], [94].
CRISPR-based gene editing is implemented with sequence-specific nucleases (SSNs) and a sgRNA to achieve precise gene knock-out (KO) or gene knock-in (KI). Additionally, researchers developed a catalytically inactive Cas9 (dCas9) that loses endonuclease activity, and has been adapted for gene expression regulation (CRISPRa/i) and 3D genome studies [33], [66], [67], [70], [84], [88]. Furthermore, base editing using modified nCas9 has greatly broadened application of the CRISPR system [32], [52], [123]. Compared with previous mature gene editing tools such as zinc-finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) [9], [10], [13], [100], [121], which use engineered proteins to target and cleave specific genome loci, CRISPR is lower in cost of both time and money. This advancing technology is increasingly being deployed, and has great potential for clinical detection, gene therapy and agricultural improvement [3], [21], [50], [126].
However, two major challenges hinder the development and application of the CRISPR/Cas system: potential off-target effects, and on-target efficiency (Fig. 1) [112], [120]. Successful CRISPR guide RNA (gRNA) design can resolve these issues [23], [96], and powerful computational approaches facilitate in silico gRNA design [19], [34], [104], thereby enabling the design of specific gRNAs for particular experiments.
In this review, we summarise existing approaches for CRIPSR guide RNA design and evaluation, and assist users in choosing favourable tools for their research. Moreover, it aims to make users aware of the latest computational CRISPR tools and resources.
2. Evaluation of CRISPR cleavage efficiency
In theory, the CRISPR/Cas protein scans the PAM sequence, and sgRNA recognises target loci and activates endonuclease activity to cleave specific sites. However, cleavage efficiency varies greatly among different target sites and/or cell lines [14], [21], [27], [28], [51], [78], [87], [98], [99], [103], [115], [117], [122], [125], suggesting that several features may influence the binding and cutting efficacy of the sgRNA-Cas complex. Numerous studies have revealed that gRNA sequence features (sequence composition, nucleotide position, GC content), genetic and epigenetic features (chromatin accessibility, gene expression) and energetics properties (RNA secondary structure, melting temperature, free energy) all contribute to gRNA efficacy. Based on these features, many computational tools have been developed for designing highly efficient gRNAs. Herein, we introduce these tools based on their features, and evaluate their efficiency (Table 1).
Table 1
Computational methods for evaluation of guide RNA efficiency.
| Tool | Enzymes | Data source | Main features | Quantitative metrics |
|---|---|---|---|---|
| E-CRISP [38] | Cas9 | – | SC, GF | – |
| CRISPRscan [78] | Cas9, Cpf1 | Zebrafish | SC | Spearman correlation = 0.309, from [36] |
| evaluateCrispr [40] | Cas9 | Drosophila | SC | Spearman correlation = 0.074, from [36] |
| sgRNAScorer [14], [15] | Cas9, Cpf1 | Human | SC, EGF | Spearman correlation = 0.225, from [36] |
| SSC [112] | Cas9 | Human, Mouse | SC | Spearman correlation = 0.274, from [36] |
| WU-CRISPR [106] | Cas9 | Human, Mouse | SC, EP | Spearman correlation = 0.215, from [36] |
| Azimuth [23], [29] | Cas9 | Human, Mouse | SC, GF, EP | Spearman correlation = 0.366, from [36] |
| CRISPRater [55] | Cas9 | Human | SC, GF | Pearson correlation = 0.399, from [55] |
| CRISPRpred [86] | Cas9 | Human, Mouse | SC, EP | ROC-AUC = 0.85, from [86] |
| CASPER [75] | Cas9, Cpf1 | – | SC | Pearson correlation = 0.443, from [75] |
| DeepCpf1 [47] | Cpf1 | Human | SC, EGF | Spearman correlation = 0.748, from [47] |
| TSAM [82] | Cas9 | Human, Mouse, Zebrafish | SC, GF, EP | Spearman correlation = 0.4, from [82] |
| TUSCAN [105] | Cas9 | Human, Mouse, Zebrafish | SC, GF | Spearman correlation = 0.12, from [105] |
| uCRISPR [119] | Cas9 | – | SC, EP | Spearman correlation = 0.3, from [119] |
Open in a separate window
2.1. Guide RNA sequence features
The nucleotide composition of a target sequence is one of the most important determinants of gRNA efficiency [24], [103], [107], [112]. Large-scale CRIPSR-based screens in mammals have shown that guanines are preferred in positions 1 and 2 before the PAM sequence [103], while thymines are disfavoured within +/-4 nucleotides surrounding the PAM sequence [107]. Additionally, sequences downstream of PAMs can influence gRNA efficiency, while sequences upstream have no significant effect [24]. Cytosine is preferred at the CRISPR/Cas9 cutting site (-3 position proximal to PAM) [21], [112], and the GC content of the region 4–13 bases downstream of the PAM sequence contributes to gRNA efficiency. Based on this key information, several efficiency prediction models have been constructed.
Rule Set 1 is a predictive model built using data derived from 1,841 sgRNAs in human and mouse [24], the score is predicted by a support vector machine (SVM) model, and a supervised learning method classifies data in a generalised linear manner. Rule Set 1 is mainly used to investigate sequence features that influence CRISPR cutting efficiency. Results predicted by this model show a high correlation with experimental results. To improve the accuracy, the authors adapted more datasets and built a new model in 2016 called Rule Set 2 [23]. In this model, position-independent nucleotide counts and the location of the sgRNA target site within the gene were considered to improve predictions based on their observations. These optimised models were applied for gRNA design for both CRISPR KO and CRISPRa/i experiments. A package to predict the gRNA efficiency based on the models was also developed and implemented in Broad Institute GPP sgRNA Designer [29].
To unravel the nucleotide preference of gRNA target sites in different CRISPR-based editing events, both CRISPR KO and CRISPRa/i libraries in mammals were screened [112], and significant differences in nucleotide preference between CRISPR KO and CRISPRa/i were detected. Elastic Net is a regularised regression method for fitting and classifying data that performs better than SVM in some cases [128]. The Elastic Net algorithm was used to construct models for both CRISPR KO and CRISPRa/i, and they were applied in Spacer Scoring for CRISPR (SSC) software to predict the efficiency of gRNA. Platforms such as E-CRISP, CHOPCHOP and CRISPR-FOCUS also include this model [12], [38], [57].
Moreno-Mateos and his colleagues observed that the loading and activity of sgRNA increased with guanine enrichment and adenine depletion [78]. They measured >1,000 sgRNAs targeting 128 genes in zebrafish and used the logistical regression method to construct a predictive model that was integrated into CRISPRscan. WU-CRISPR takes advantage of this data and adds some novel features [24], [106], resulting in a model with higher precision than several other predictive models [14], [24], [112]. Labuhn et al. identified PAM-distal GC content-dependent activity and constructed a model named CRISPRater [55] that was integrated into CRISPR/Cas9 target online predictor (CCTop), a platform for CRISPR target prediction [93].
The Church laboratory developed software called sgRNA scorer to calculate sgRNA on-target scores based on their SVM model [14]. A second version of the sgRNA scorer software was proposed that improved the on-target prediction power and added prediction for other Cas systems such as SaCas9 and AsCpf1 [15]. Housden et al. then used a drug target method to screen CRISPR KO efficiency in Drosophila and developed an efficiency prediction tool [40]. CASPER integrated scores from CRISPRscan and added some new features to maximise correlations between scores and on-target experimental data [75]. This tool can also detect off-target scores and perform multipopulational analysis.
2.2. Genetic and epigenetic features
Genetic and epigenetic features like chromatin accessibility, gene position and expression are also important factors that influence sgRNA binding and Cas nucleases cleavage. Many researches have demonstrated that nucleosomes inhibit Cas9 target cleavage, and DNase I hypersensitivity and epigenomic markers alter gRNA efficacy [23], [39], [44], [101], [116]. Based on these features, several tools have been developed. By borrowing knowledge from oligonucleotide design and nucleosome occupancy models, an R package called predictSGRNA was proposed for evaluation of sgRNA efficacy [53], and this performed better than other models such as Azimuth and sgRNA scorer [14], [23], [29].
Non-homologous end joining (NHEJ) and microhomology-mediated end joining (MMEJ) are two major pathways which produce heterogeneous repair outcomes when repairing Cas9-mediated DSBs. People use CRISPR/Cas9 system to knock out genes by inducing indels into target genome location. However, CRISPR-based gene KO may induces in-frame variants in which gene functions are retained. Thus, microhomology-based prediction of CRISPR on-target efficiency should be considered. To this end, Bae et al. developed Microhomology-Predictor to improve KO efficiency by reducing in-frame editing [7]. Recent researches have showed that template-free Cas9-editing outcomes are predictable. inDelphi was the first model for precise prediction of CRISPR editing genotype [90]. Soon after, a computational predictor called FORECasT was developed using >40,000 sgRNAs in different cell lines [5]. It was shown that most reproducible mutations are single base insertion, short deletions or longer microhomology-mediated deletions, in addition, Cas9-editing outcomes were cell-line-dependent. The Shendure laboratory also built a predictive model called Lindel for prediction of the insertions and deletions of CRISPR/Cas9-mediated DSB repair based on local sequence context [16]. All these approaches are sure to assist users in guide RNAs selection for gene disruption.
CRISPR-Cpf1 achieves high efficiency and suffers minor off-targets, besides, Cpf1 prefers AT-enriched regions. Hence, more and more studies have adapted Cpf1 for KO screens. However, models for evaluation of Cpf1 cleavage efficiency are lacking. DeepCpf1 is an algorithm especially for prediction of Cpf1 activity [47]. It is implemented within the deep-learning framework and chromatin accessibility data. This program can significantly improve the accuracy of Cpf1 activity prediction. In addition, models for other Cas experiments, such as Cas9, xCas9, and base edition are also provided on the author’s GitHub (https://github.com/MyungjaeSong/Paired-Library). CRISPR-DT is a recently developed platform for prediction of Cpf1 target efficiency [127]. This SVM model displayed better performance than the deep learning-based model employed in DeepCpf1.
2.3. Energetics properties
The energetics associated with the formation of the DNA, gRNA and Cas protein complex are regular and can be analysed to eliminate bias among different models, since some energetics methods may better illustrate the Cas9 editing efficacy [95], [107], [113]. CRISPRpred includes the positions of nucleotides as well as secondary structures of sgRNAs to predict the cleavage efficiency, and this performs better than Rule Set 1 [24], [86]. Zhang et al. recently demonstrated a free energy scheme called uCRISPR for evaluating the Cas9 editing efficacy, as well as off-target effects [119]. This model is thought to apply to any cleavage-activity Cas9 dataset.
2.4. Other considerations for gRNA efficiency
Of note, these predictive models were trained by individual experiments and rules, and each model generates different on-target scores for sgRNAs, hence users must be careful when evaluating or designing guide RNAs using these models in their own experiments. However, several key features are reliable, such as guanine preference, GC content, seed region and the secondary structure of gRNAs [65], [91], [106], [112].
The accuracy of different models is controlled by their learning methods or the approaches of CRISPR activity measurement. Doench et al. tested multiple training methods and selected the best-performing one as the kernel of their model [23]. TUSCAN, a random forest-based model, outperformed models built solely by linear regression [78], [105]. A two-step averaging method (TSAM) for the regression of cleavage efficiencies also performed better than many other models [14], [23], [82], [112]. Additionally, measurement using sequencing data rather than phenotypic data may generate fewer false positive results, albeit at a cost [105].
It is critical for users to know which tool best suits their research. A comprehensive evaluation of different efficiency prediction tools was conducted to examine differences among models [36], and it revealed differences in the correlation between different datasets and models. Furthermore, no model performed excellently across all datasets, suggesting that a careful selection of CRISPR gRNA design tools is necessary. Users can also evaluate gRNAs using multiple models and select the best one for their experiments. Among the available tools, Rule Set 2 and DeepCpf1 are the most used and accurate scoring methods for evaluating Cas9 and Cpf1 cutting efficacy, and uCRISPR may be more accurate than some other methods, but it requires further experimental testing.
3. Prediction of CRISPR cutting specificity
The main obstacle for the application of CRISPR is off-target effects. CRISPR nucleases may cleave unintended genomic sites and cause unexpected mutations due to sgRNAs recognising DNA sequences with a few mismatches and/or DNA/RNA bulges, referred to as off-target cleavage [41], [120]. Off-target effects can be effectively relieved by predicting CRISPR cutting specificity and designing optimal gRNAs [41]. To predict the specificity of CRISPR gRNAs, two main methods have been proposed: (1) alignment-based method. Based on conventional or specialized algothrims, gRNAs are aligned to a given genome and off-target sequences and sites are returned. This method is mainly used for find out all potential off-targets in silico, (2) Scoring-based method. sgRNAs should be further scoring and ranking using identified off-targets from alignment process to select the most specific one for experiments. Two scoring approaches are shown: hypothesis-driven, where off-targets are scored based on the contribution of specific genome context factors to gRNA specificity; learning-based, where gRNAs are scored and predicted from a training model that considers the different features affecting specificity. These methods for prediction of gRNA specificity are discussed below and some of them are summarised in Table 2.
Table 2
Computational methods for prediction of guide RNA specificity.
| Tool | Enzymes | Methods | Main features | Quantitative metrics |
|---|---|---|---|---|
| CasOT [108] | Cas9 | alignment | unlimited mismatch number, paired-gRNA mode, annotation | slow |
| Cas-OFFinder [8] | costom | alignment | unlimited mismatch number, GPU acceleration, web support | middle, fast (use GPU) |
| sgRNAcas9 [110] | Cas9 | alignment | max 5 mismatches, paired-gRNA mode, annotation, risk evaluation | slow |
| FlashFry [74] | costom | alignment | unlimited mismatch number, multiple on/off-target scores, annotation | fast |
| Crisflash [43] | Cas9 | alignment | unlimited mismatch number, variant data support | fast |
| MIT [41] | Cas9 | scoring | 20 bp sgRNA without PAM | ROC-AUC = 0.87, from [36] |
| CCTop [93] | Cas9, Cpf1 | scoring | empirically score based on number of mismatches | ROC-AUC = 0.77, from [36] |
| CFD [23] | Cas9 | scoring | 20 bp sgRNA with PAM (enable non-canonical PAM) | ROC-AUC = 0.91, from [36] |
| CRISPRoff [4] | Cas9 | scoring | energetics property and sequences composition | ROC-AUC = 0.98, from [4] |
| uCRISPR [119] | Cas9 | scoring | energetics property and sequences composition | Pearson correlation = 0.75, from [119] |
| CRISTA [1] | Cas9 | scoring | machine learning, sequences composition and epigenetic feature | ROC-AUC = 0.96, from [1] |
| Elevation [62] | Cas9 | scoring | machine learning, integrate both CFD model and epigenetic features | ROC-AUC = 0.98, from [62] |
| DeepCRISPR [18] | Cas9 | scoring | deep learning, sequences composition and epigenetic feature | ROC-AUC = 0.98, from [18] |
Open in a separate window
3.1. Alignment-based methods
In theory, potential off-target sites can be identified by aligning gRNA sequences to the reference genome based on sequence homology. Bowtie [59] and BWA [61] are traditional tools for short sequence alignment that are capable of off-target detection [36], [104]. However, there are several potential issues when using these tools. First, tools like Bowtie and BWA cannot identify small PAMs, since these alignment tools were developed for next-generation sequencing (NGS) read alignment. Second, these tools allow very limited mismatches in their seed regions, making alignment with default parameters impractical for identifying all potential off-target sites. A survey using bowtie2 [58] to detect off-targets failed to find all possible off-target sites, and could only find off-targets with up to one mismatch [23].
Despite the defects, some gRNA design methods utilise these tools, and parameters have been modified to fit the demand for off-target prediction. CCTop uses Bowtie to find off-target sites by first identifying PAM sites, and matches and mismatches in protospacer sequences are then searched for by bowtie with modified parameters [93]. Up to five mismatches are allowed in the protospacer as more mismatches may prevent DSB induction. CCTop also implements an off-target score for each candidate sgRNA. CHOPCHOP detects off-target sites using Bowtie with parameters -v and -L for searching sgRNA core regions [77]. CRISPOR uses BWA to find all potential off-target sites in iterative mode (“-N”), and can find all validated off-targets as well as Cas-OFFinder [8], [36].
In addition to these traditional tools, many other tools and algorithms have been developed for off-target site detection. Cas-OFFinder is one of the most popular tools for searching potential off-target sites, and advantages include no limit to the number of mismatches, PAM types, gRNA length or high running speed with GPUs. Cas-OFFinder can also predict off-target sites with 1 bp deletions or insertions (i.e. DNA/RNA bulges). CasOT is implemented to find Cas9 on-target sites from input sequences, as well as potential off-target sites with up to six mismatches in the seed region (12 nt adjacent to the PAM). This tool can also determine whether off-targets are within a coding exon [108]. Meanwhile, sgRNAcas9 utilises the ultrafast short sequence mapping tool SeqMap [45] to find off-targets, and classifies all sites into three categories to generate a final output of the best candidate gRNAs [110]. Recently, two new alignment-based tools have been developed. Crisflash utilises a tree-based algorithm to rapidly design CRISPR guide RNAs and optimise guide accuracy by incorporating user-supplied variant data [43]. FlashFry rapidly searches off-target sites and provides much useful information (annotation of off-target sites, on/off-target scores, GC content, etc.) for candidate gRNAs [74]. Here we still classified Crisflash and FlashFry as alignment-based methods since they both propose novel algorithms for off-target searching, whereas the scoring approaches they use are borrowed from others [23], [24], [41], [78].
Among these tools, Cas-OFFinder may be the best choice for identifying all potential off-target sites with any Cas nucleases, and FlashFry is also worth a try for its high speed and comprehensive outputs.
3.2. Scoring-based methods
3.2.1. Hypothesis-driven methods
Alignment-based methods are reliable for detection of most potential off-targets, however, not all nucleotide positions containing mismatches have the same decisive effect on off-target cleavage. Additionally, alignment-based prediction always outputs redundant off-target sites, many which are false-positives, although users can reduce the number of outputs by restricting the maximum mismatches when exploring off-target cleavage. One study compared experimentally validated off-targets and off-targets predicted by Cas-OFFinder and CCTop, and the results showed that off-targets detected by the prediction tools only covered some of the validated sites, while some off-target sites cannot be predicted solely based on sequence homology [11]. Thus, features that influence the nonspecific binding of CRISPR gRNAs need to be considered to increase the accuracy of off-target detection.
MIT (Hsu-Zhang) score was proposed for off-target evaluation during the early stages of gene editing by CRISPR. Hsu et al. evaluated >700 guide RNA variants and SpCas9-induced indel mutation levels at >100 predicted genomic off-target loci [41]. They evaluated the contributions made by different mismatch positions and numbers in the target 20 bp, and calculated a weight matrix to determine off-target efficiency for each sgRNA. The authors claimed that this score accounts for >50% of the variance in cutting frequency. The MIT score has been integrated into many CRISPR gRNA design tools such as CHOPCHOP and CRISPOR [36], [56].
Cutting frequency determination (CFD) is another popular score for off-target evaluation. It is noticeable that sgRNA can also bind genome loci with non-canonical PAMs such as NAG, NCG and NGA, which may cause off-target cleavage. Doench et al. added PAM features in their scoring metrics [23]. sgRNAs with mismatches and indels in target sequences were also included for examining correlations between sgRNAs and off-targets. CFD score was validated with GUIDE-seq and proved to perform better than MIT score, hence it was adopted by CRISPRscan [78], GuideScan [83] and CRISPOR [36].
The prediction of sgRNA specificity based on the structural features of the Cas9-sgRNA complex proved to be superior to prediction solely based on sequence features. CRISPRoff [4] and uCRISPR [119] both utilise energetics properties for off-target evaluation. Compared with other scoring methods like MIT and CFD, they both yielded better accuracy in off-target prediction. Nevertheless, neither have not been systematically evaluated by large-scale experiments, and caution should be exercised when using them.
3.2.2. Learning-based methods
Sometimes, empirical algorithms cannot effectively evaluate off-targets since they only consider minor features, whereas learning-based methods build complex models using combinations of multiple features, and they may better predict off-targets. In recent years, several new approaches for off-target prediction based on machine learning have been developed.
CRISPR Target Assessment (CRISTA) software uses BWA as the off-target search tool and implements multiple features (PAM type, nucleotide composition, GC content, chromatin structure, DNA methylation, RNA secondary structure, etc.) to predict cleavage propensity [1]. CRISTA exhibits better performance than MIT, CCTop and CFD.
The Doench laboratory also developed a linear regression model for prediction of off-target activity called Elevation that takes both sequence and chromatin accessibility features into consideration, as well as observations from others [62]. Elevation predicts individual scores for each off-target site, as well as an aggregate score for gRNAs. This method performs best among MIT, CFD and CCTop. However, this software can only compute off-target scores for the human exome (GRCh38) and cannot support other organisms on their website.
DeepCRISPR is a state-of-the-art computational platform that unifies sgRNA on-target and off-target site prediction into one framework with deep learning [18]. It identifies all possible sequence and epigenetic features that may affect sgRNA KO efficacy by learning from large data. This method was proved to surpass other available off-target prediction tools [23], [41], [92], [93].
3.3. Benchmark of scoring-based methods
Different scoring methods are based on different characteristics applied by each individual laboratory. In order to compare the accuracy of different scores, Haeussler et al. assessed four hypothesis-driven methods for off-target prediction using the same datasets [36]. CFD score exhibited the best prediction accuracy, whereas CCTop performed the worst. Data imbalance, where the number of true observed off-target sites (OTS) identified by off-target detection experiments is much less than that of all potential off-target sites recognized by alignment-based methods, is a common issue in the learning-based methods. To address the problem, a systematic survey was conducted to assess the influence of data imbalance [30]. The authors used bootstrapping sampling and PR-AUC methods to evaluate two well-established models. They emphasized the importance of using symmetric data for model construct and taking unbiased datasets for benchmark. According to current assessment results, we recommend people to use CFD score for off-target prediction. Elevation and DeepCRISPR are suitable for human genome editing, but CRISPRoff and uCRISPR may need further evaluation before use.
4. Tools for CRISPR guide RNA design
A CRISPR/Cas complex typically contains a Cas protein and a sgRNA, both of which determine the cutting activity. However, protein engineering is a complicated and uncertain strategy for most researchers, and optimising gRNAs is more feasible and efficient. A robust gRNA requires maximum on-target efficiency as well as minimum off-target activity. Basing on these two criteria, numerous computational tools have been developed for high-efficiency CRISPR gRNA design. However, each tool possesses distinct features, and user-friendliness is also important. To acquaint users with existing gRNA design tools, we provide an overview of most that are available based on the strength of their features, and this help users to make appropriate selections. Furthermore, we recommend several functional and user-friendly websites for gRNA design (Fig. 2,Table 3).
4.1. A comprehensive overview of CRISPR guide RNA design tools
The CRISPR/Cas system was first adapted for gene editing in 2012, and several tools were developed immediately thereafter, such as Zinc Finger Targeter (ZiFiT) [89], Cas9 guide RNA Design [68] and CRISPR (http://crispr.mit.edu/) [41]. All these platforms have been implemented in the design of gRNAs for model organisms. ZiFiT is also used to design Zinc Finger and TALEN modules, but Cas9 guide RNA Design and CRISPR are now unavailable.
In the following years, various tools for CRISPR gRNA design emerged, due to both urgent demand for these tools, and because people have learned more and more about how the CRISPR system functions and what influences the efficiency and specificity of Cas cleavage. Many of these tools combine multiple scoring methods and provide alternative options for users. Some others have also proposed their own algorithms to rank designed sgRNAs, and this can assist users in gRNA selection. CHOPCHOP [56], [57], [77] provides alternative scores for users such as Rule Set ½ [23], [24], SSC [112], CRISPRscan [78] and deepCpf1 [47]. E-CRISP utilises its own SAE (Specificity, Annotation, Efficacy) score to determine the quality of each sgRNA, while Rule Set 1 [24] and SSC [112] are also included in E-CRISP. CCTop [93] assigns off-target scores for each sequence empirically, while the CRISPRater score [55] is also included to predict the efficiency of sgRNAs. CRISPOR [36] is a versatile platform that ranks gRNAs according to different scores for evaluating potential off-targets in the genome of interest, and for predicting on–target activity.
A large number of CRISPR/Cas-derived RNA-guided Endonucleases (RGENs) have been identified or modified to improve the cutting efficiency and enlarge the editing range. Some tools enable the design of gRNAs for RGENs. For example, Cas-Designer [79] allows users to choose 20 PAM types from different RGENs, while CRISPOR [20] also offers various PAMs from a defined list. To best serve the specialised purposes of different experiments, several web-based tools have been developed. CRISPR-ERA [64] and CHOPCHOP v3 [57] support sgRNA design for the CRISPRa/i system. CRISPETa [85] is mainly used for genome deletion with paired gRNAs, and BE-Designer [42] can be used for designing gRNA for base editing. Recently, three methods have been employed for successively predicting Cas9-editing outcomes: inDelphi [90], FORECasT [5] and Lindel [16]. These approaches can help to identify gRNAs based on forecasting results.
Researchers should choose suitable tools with caution since different tools are in favour of different genomes or cell types. For instance, Yeastriction [72] is specialised for yeast, FlyCRISPR [35] is specific for Drosophila, EuPaGDT [81] is recommended for eukaryotic pathogens, and CRISPR-P [60], [63], CRISPR-PLANT [76], [109] and CRISPR-GE [111] are all implemented for genome editing in plants. Also, user-friendliness is essential for these web-based design tools. Based on these considerations, we propose three comprehensive web-based platforms for CRISPR gRNA design.
4.2. Three comprehensive web platforms for CRISPR guide RNA design
4.2.1. CHOPCHOP
CHOPCHOP has a succinct interface with well-rounded functions, >200 genomes are available on the website, and users can provide gene names, genomic coordinates or target sequences as inputs. If a gene is given, users can set specific regions of the gene as targets, such as coding sequences or promoters. Before designing gRNAs, two off-target detection methods and seven efficiency scores can be chosen, which aids users in selecting optimal gRNAs for their research.
To satisfy different experimental purposes, CRISPR Cas9 nuclease/nickase, Cpf1, CasX, C2C2 and TALEN are all supported by CHOPCHOP, and various modes of DNA targeting are optional such as KO/KI, gene activation/repression, and nanopore enrichment. In activation/repression mode, gRNAs are designed to target the promoter region and its flanking sites in order to bring the activating/repressing domain into close proximity with the transcription start site [17], [54]. Meanwhile, nanopore enrichment mode is implemented to design sgRNAs for long fragment excision within 40 kb. Additionally, the inDelphi model has been integrated into CHOPCHOP for repair profile prediction by Cas9 in five cell types [90].
After clicking the ‘Find Target Sites’ button, a results table is shown, and each candidate gRNA has a rank, genomic information, GC content, self-complementarity score, mismatch number (0–3) and an efficiency score. A graphical view of all gRNAs is also provided in the UCSC Genome Browser, which helps users to select optimal gRNAs.
4.2.2. CRISPR RGEN tools
CRISPR RGEN tools, computational tools and libraries for RNA-guided endonucleases, are maintained by the Bae laboratory, and libraries comprise nine useful tools including Cas-OFFinder [8], Cas-Designer [79] and Digenome-Seq [80]. Herein, we mainly discuss the two gRNA design tools Cas-Designer and BE-Designer [42].
Compared with other designer tools, Cas-Designer allows DNA/RNA bulges when performing off-target detection. Additionally, this detection method is more rapid than others due to the Cas-OFFinder algorithm. Genomic sequences or coordinates and fasta file formats are allowed as inputs. Over 350 genomes and 20 PAM types are specified for users, and outputs include target sequences as well as out-of-frame score (calculate by microhomology), mismatch number (0–2) and off-target sites with up to 1 bp bulge. On/off-target sites are redirected to the Ensembl genome browser [26], which is capable of further inspection.
Unlike Cas-Designer, BE-Designer is primarily implemented for base editing. In this tool, four methods of base editing are specified, and the editing window region is also adjustable. After the design phase, three outcomes are based on different coding types, and gRNAs, editing window sequences and amino acids are highlighted in a user-friendly manner. This tool is a good choice for base editing.
4.2.3. CRISPOR
Since many tools have been developed for highly efficient CRISPR gRNA design, an ensemble of multiple tools can be useful, and CRISPOR does this well [20], [36]. At present, CRISPOR contains 417 genomes and 19 PAM types, and can meet the needs of most users. CRISPOR takes genome coordinates and sequences <2,000 bp as inputs and provides comprehensive information as outputs. By default, results are shown in two parts; visualisation of PAM sites along the given sequence, which allows for downloading using multiple formats including fasta, GenBank and SnapGene; a table including all information for each predicted gRNA is also generated. In the table, two specificity scores (MIT and CFD) and 10 efficiency scores (Rule Set 2, CRISPRscan, etc.) are calculated [23], [41], [78]. Furthermore, Microhomology and Lindel scores are also provided for outcome prediction [7], [16]. Warnings such as extreme GC content and poly-T values are indicated, the table is downloadable, and candidate off-target sites with up to four mismatches can be visualised and downloaded.
In addition to gRNA design, CRISPOR designs primers for each gRNA as well as off-target sites. These primers are useful when conducting on/off-target validation or in vitro expression experiments. Furthermore, CRISPOR enables filtering of gRNAs with genomic variants based on well-known variant databases or marked input sequences with N characters. Thus, CRISPOR may be the best tool for designing gRNAs.
5. Summary and perspectives
CRISPR/Cas technology has emerged as an advanced strategy for functional genomics, crop breeding and precise medicine. Guide RNAs are indispensable for CRISPR-based editing, and computational approaches provide assistance for efficient gRNA design. Numerous tools have been developed for CRISPR gRNA design and evaluation. However, many issues remain. For example, experimental datasets used to build models for predicting sgRNA specificity or efficiency are disparate; CRISPRscan score performs worse in mammals than in zebrafish, the genome the predictive model was trained in [36]. Researchers should therefore know the strengths and weaknesses of each scoring method, and select the optimal tool for their research.
As we become more aware of the mechanisms by which Cas proteins recognise gRNAs, and bind to and cleave target loci, more and more novel features contributing to cutting efficiency and specificity are being identified, including sequence composition. For instance, low chromatin accessibility may block access of Cas9, while open chromatin regions are more likely to cause undesired mutations [39], [92]. In practice, specificity is more important than efficiency for application of CRISPR. However, several off-targets remain indecipherable using current tools, and discovery of novel features may minimise off-target effects.
Too much choice is not always desirable, and an unbiased evaluation can provide guidance. To this end, the Liu laboratory has conducted the benchmarking [30], [114], and Haeussler et al. performed a comparison of different predictive scores and integrated most into CRISPOR [36]. None of the tools excel when using heterogeneous data, due to cell-specific or species-specific training models. Therefore, more cell types need to be evaluated. Also, plant and some animal genomes should be analysed thoroughly, since human or mouse have dominated to date. Moreover, it will be beneficial if useful features of multiple tools are combined in future software packages.
The major outcomes of Cas9 cleavage is non-random and predictable [102], and several tools have been created for prediction of CRISPR-Cas9 outcomes [5], [16], [90]. Such findings facilitate more accurate gene editing. However, existing tools cannot account for large indels, homozygous sites and the activity of nucleases other than SpCas9, and this should be resolved in future [6].
In summary, the development of computational approaches has revolutionised the design of effective gRNAs, and CRISPR-based gene therapies and customised gene editing may come within reach. It is likely that CRISPR technology will continue to become more powerful in the coming years.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by the National Transgenic Major Project (2019ZX08010003-001-002, 2018ZX08020-003), Jiangsu Province Government Project (BK2018003)/The open funds of the Jiangsu Key Laboratory of Crop Genomics and Molecular Breeding (PL201801) for T.Z. and Y.Z.; the Fund of Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) for T.Z.
References
1. Abadi S., Yan W.X., Amar D., Mayrose I. A machine learning approach for predicting CRISPR-Cas9 cleavage efficiencies and patterns underlying its mechanism of action. PLoS Comput Biol. 2017;13[PMC free article] [PubMed] [Google Scholar]
2. Abudayyeh O.O., Gootenberg J.S., Essletzbichler P., Han S., Joung J., Belanto J.J. RNA targeting with CRISPR-Cas13. Nature. 2017;550:280–284.[PMC free article] [PubMed] [Google Scholar]
3. Adli M. The CRISPR tool kit for genome editing and beyond. Nat Commun. 2018:9.[PMC free article] [PubMed] [Google Scholar]
4. Alkan F., Wenzel A., Anthon C., Havgaard J.H., Gorodkin J. CRISPR-Cas9 off-targeting assessment with nucleic acid duplex energy parameters. Genome Biol. 2018;19[PMC free article] [PubMed] [Google Scholar]
5. Allen F., Crepaldi L., Alsinet C., Strong A.J., Kleshchevnikov V., De Angeli P. Predicting the mutations generated by repair of Cas9-induced double-strand breaks. Nat Biotechnol. 2019;37:64–72.[PMC free article] [PubMed] [Google Scholar]
6. Bae S., Kim J.S. Machine learning finds Cas9-edited genotypes. Nat Biomed Eng. 2018;2:892–893. [PubMed] [Google Scholar]
7. Bae S., Kweon J., Kim H.S., Kim J.S. Microhomology-based choice of Cas9 nuclease target sites. Nat Methods. 2014;11:705–706. [PubMed] [Google Scholar]
8. Bae S., Park J., Kim J.S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30:1473–1475.[PMC free article] [PubMed] [Google Scholar]
9. Boch J., Scholze H., Schornack S., Landgraf A., Hahn S., Kay S. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009;326:1509–1512. [PubMed] [Google Scholar]
10. Bogdanove A.J., Voytas D.F. TAL effectors: customizable proteins for DNA targeting. Science. 2011;333:1843–1846. [PubMed] [Google Scholar]
11. Cameron P., Fuller C.K., Donohoue P.D., Jones B.N., Thompson M.S., Carter M.M. Mapping the genomic landscape of CRISPR-Cas9 cleavage. Nat Methods. 2017;14:600–606. [PubMed] [Google Scholar]
12. Cao Q., Ma J., Chen C.H., Xu H., Chen Z., Li W. CRISPR-FOCUS: a web server for designing focused CRISPR screening experiments. PLoS ONE. 2017;12[PMC free article] [PubMed] [Google Scholar]
13. Carroll D. Genome engineering with zinc-finger nucleases. Genetics. 2011;188:773–782.[PMC free article] [PubMed] [Google Scholar]
14. Chari R., Mali P., Moosburner M., Church G.M. Unraveling CRISPR-Cas9 genome engineering parameters via a library-on-library approach. Nat Methods. 2015;12:823–826.[PMC free article] [PubMed] [Google Scholar]
15. Chari R., Yeo N.C., Chavez A., Church G.M. sgRNA Scorer 2.0: a species-independent model to predict CRISPR/Cas9 activity. ACS Synth Biol. 2017;6:902–904.[PMC free article] [PubMed] [Google Scholar]
16. Chen W., McKenna A., Schreiber J., Haeussler M., Yin Y., Agarwal V. Massively parallel profiling and predictive modeling of the outcomes of CRISPR/Cas9-mediated double-strand break repair. Nucleic Acids Res. 2019;47:7989–8003.[PMC free article] [PubMed] [Google Scholar]
17. Cheng A.W., Wang H., Yang H., Shi L., Katz Y., Theunissen T.W. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res. 2013;23:1163–1171.[PMC free article] [PubMed] [Google Scholar]
18. Chuai G.H., Ma H.H., Yan J.F., Chen M., Hong N.F., Xue D.Y. DeepCRISPR: optimized CRISPR guide RNA design by deep learning. Genome Biol. 2018;19[PMC free article] [PubMed] [Google Scholar]
19. Chuai G.H., Wang Q.L., Liu Q. In silico meets in vivo: towards computational CRISPR-based sgRNA design. Trends Biotechnol. 2017;35:12–21. [PubMed] [Google Scholar]
20. Concordet J.P., Haeussler M. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018;46:W242–W245.[PMC free article] [PubMed] [Google Scholar]
21. Cong L., Ran F.A., Cox D., Lin S.L., Barretto R., Habib N. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823.[PMC free article] [PubMed] [Google Scholar]
22. Deltcheva E., Chylinski K., Sharma C.M., Gonzales K., Chao Y.J., Pirzada Z.A. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471:602–607.[PMC free article] [PubMed] [Google Scholar]
23. Doench J.G., Fusi N., Sullender M., Hegde M., Vaimberg E.W., Donovan K.F. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol. 2016;34:184–191.[PMC free article] [PubMed] [Google Scholar]
24. Doench J.G., Hartenian E., Graham D.B., Tothova Z., Hegde M., Smith I. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat Biotechnol. 2014;32:1262–1267.[PMC free article] [PubMed] [Google Scholar]
25. Endo A., Masafumi M., Kaya H., Toki S. Efficient targeted mutagenesis of rice and tobacco genomes using Cpf1 from Francisella novicida. Sci Rep-Uk. 2016:6.[PMC free article] [PubMed] [Google Scholar]
26. Flicek P., Amode M.R., Barrell D., Beal K., Brent S., Chen Y. Ensembl 2011. Nucleic Acids Res. 2011;39:D800–D806.[PMC free article] [PubMed] [Google Scholar]
27. Fu Y.F., Foden J.A., Khayter C., Maeder M.L., Reyon D., Joung J.K. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013;31:822–826.[PMC free article] [PubMed] [Google Scholar]
28. Fu Y.F., Sander J.D., Reyon D., Cascio V.M., Joung J.K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol. 2014;32:279–284.[PMC free article] [PubMed] [Google Scholar]
29. Fusi N., Smith I., Doench J., Listgarten J. In silico predictive modeling of CRISPR/Cas9 guide efficiency. bioRxiv. 2015[Google Scholar]
30. Gao Y., Chuai G., Yu W., Qu S., Liu Q. Data imbalance in CRISPR off-target prediction. Brief Bioinform. 2019:bbz069. [PubMed] [Google Scholar]
31. Gasiunas G., Barrangou R., Horvath P., Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. P Natl Acad Sci USA. 2012;109:E2579–E2586.[PMC free article] [PubMed] [Google Scholar]
32. Gaudelli N.M., Komor A.C., Rees H.A., Packer M.S., Badran A.H., Bryson D.I. Programmable base editing of A.T to G.C in genomic DNA without DNA cleavage. Nature. 2017;551:464–471.[PMC free article] [PubMed] [Google Scholar]
33. Gilbert L.A., Horlbeck M.A., Adamson B., Villalta J.E., Chen Y., Whitehead E.H. Genome-scale CRISPR-mediated control of gene repression and activation. Cell. 2014;159:647–661.[PMC free article] [PubMed] [Google Scholar]
34. Graham D.B., Root D.E. Resources for the design of CRISPR gene editing experiments. Genome Biol. 2015;16[PMC free article] [PubMed] [Google Scholar]
35. Gratz S.J., Ukken F.P., Rubinstein C.D., Thiede G., Donohue L.K., Cummings A.M. Highly specific and efficient CRISPR/Cas9-catalyzed homology-directed repair in Drosophila. Genetics. 2014;196:961–971.[PMC free article] [PubMed] [Google Scholar]
36. Haeussler M., Schonig K., Eckert H., Eschstruth A., Mianne J., Renaud J.B. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016;17[PMC free article] [PubMed] [Google Scholar]
37. Harrington L.B., Burstein D., Chen J.S., Paez-Espino D., Ma E., Witte I.P. Programmed DNA destruction by miniature CRISPR-Cas14 enzymes. Science. 2018;362:839–842.[PMC free article] [PubMed] [Google Scholar]
38. Heigwer F., Kerr G., Boutros M. E-CRISP: fast CRISPR target site identification. Nat Meth. 2014;11:122–124. [PubMed] [Google Scholar]
39. Horlbeck M.A., Witkowsky L.B., Guglielmi B., Replogle J.M., Gilbert L.A., Villalta J.E. Nucleosomes impede Cas9 access to DNA in vivo and in vitro. Elife. 2016;5[PMC free article] [PubMed] [Google Scholar]
40. Housden B.E., Valvezan A.J., Kelley C., Sopko R., Hu Y., Roesel C. Identification of potential drug targets for tuberous sclerosis complex by synthetic screens combining CRISPR-based knockouts with RNAi. Sci Signal. 2015:8.[PMC free article] [PubMed] [Google Scholar]
41. Hsu P.D., Scott D.A., Weinstein J.A., Ran F.A., Konermann S., Agarwala V. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31:827–832.[PMC free article] [PubMed] [Google Scholar]
42. Hwang G.H., Park J., Lim K., Kim S., Yu J., Yu E. Web-based design and analysis tools for CRISPR base editing. BMC Bioinf. 2018;19:542.[PMC free article] [PubMed] [Google Scholar]
43. Jacquin A.L., Odom D.T., Lukk M. Crisflash: open-source software to generate CRISPR guide RNAs against genomes annotated with individual variation. Bioinformatics. 2019;35:3146–3147.[PMC free article] [PubMed] [Google Scholar]
44. Jensen K.T., Floe L., Petersen T.S., Huang J., Xu F., Bolund L. Chromatin accessibility and guide sequence secondary structure affect CRISPR-Cas9 gene editing efficiency. FEBS Lett. 2017;591:1892–1901. [PubMed] [Google Scholar]
45. Jiang H., Wong W.H. SeqMap: mapping massive amount of oligonucleotides to the genome. Bioinformatics. 2008;24:2395–2396.[PMC free article] [PubMed] [Google Scholar]
46. Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821.[PMC free article] [PubMed] [Google Scholar]
47. Kim H.K., Min S., Song M., Jung S., Choi J.W., Kim Y. Deep learning improves prediction of CRISPR-Cpf1 guide RNA activity. Nat Biotechnol. 2018;36:239–241. [PubMed] [Google Scholar]
48. Kim Y., Cheong S.A., Lee J.G., Lee S.W., Lee M.S., Baek I.J. Generation of knockout mice by Cpf1-mediated gene targeting. Nat Biotechnol. 2016;34:808–810. [PubMed] [Google Scholar]
49. Kleinstiver B.P., Tsai S.Q., Prew M.S., Nguyen N.T., Welch M.M., Lopez J.M. Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells. Nat Biotechnol. 2016;34:869–874.[PMC free article] [PubMed] [Google Scholar]
50. Knott G.J., Doudna J.A. CRISPR-Cas guides the future of genetic engineering. Science. 2018;361:866–869.[PMC free article] [PubMed] [Google Scholar]
51. Koike-Yusa H., Li Y.L., Tan E.P., Velasco-Herrera M.D., Yusa K. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat Biotechnol. 2014;32:267–273. [PubMed] [Google Scholar]
52. Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424.[PMC free article] [PubMed] [Google Scholar]
53. Kuan P.F., Powers S., He S., Li K., Zhao X., Huang B. A systematic evaluation of nucleotide properties for CRISPR sgRNA design. BMC Bioinf. 2017;18[PMC free article] [PubMed] [Google Scholar]
54. La Russa M.F., Qi L.S. The New State of the Art: Cas9 for gene activation and repression. Mol Cell Biol. 2015;35:3800–3809.[PMC free article] [PubMed] [Google Scholar]
55. Labuhn M., Adams F.F., Ng M., Knoess S., Schambach A., Charpentier E.M. Refined sgRNA efficacy prediction improves large- and small-scale CRISPR-Cas9 applications. Nucleic Acids Res. 2018;46:1375–1385.[PMC free article] [PubMed] [Google Scholar]
56. Labun K., Montague T.G., Gagnon J.A., Thyme S.B., Valen E. CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Res. 2016;44:W272–W276.[PMC free article] [PubMed] [Google Scholar]
57. Labun K., Montague T.G., Krause M., Torres Cleuren Y.N., Tjeldnes H., Valen E. CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 2019;47:W171–W174.[PMC free article] [PubMed] [Google Scholar]
58. Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359.[PMC free article] [PubMed] [Google Scholar]
59. Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10[PMC free article] [PubMed] [Google Scholar]
60. Lei Y., Lu L., Liu H.Y., Li S., Xing F., Chen L.L. CRISPR-P: a web tool for synthetic single-guide RNA design of CRISPR-system in plants. Mol Plant. 2014;7:1494–1496. [PubMed] [Google Scholar]
61. Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760.[PMC free article] [PubMed] [Google Scholar]
62. Listgarten J., Weinstein M., Kleinstiver B.P., Sousa A.A., Joung J.K., Crawford J. Prediction of off-target activities for the end-to-end design of CRISPR guide RNAs. Nat Biomed Eng. 2018;2:38–47.[PMC free article] [PubMed] [Google Scholar]
63. Liu H., Ding Y., Zhou Y., Jin W., Xie K., Chen L.L. CRISPR-P 2.0: an improved CRISPR-Cas9 tool for genome editing in plants. Mol Plant. 2017;10:530–532. [PubMed] [Google Scholar]
64. Liu H.L., Wei Z., Dominguez A., Li Y.D., Wang X.W., Qi L.S. CRISPR-ERA: a comprehensive design tool for CRISPR-mediated gene editing, repression and activation. Bioinformatics. 2015;31:3676–3678.[PMC free article] [PubMed] [Google Scholar]
65. Liu X., Homma A., Sayadi J., Yang S., Ohashi J., Takumi T. Sequence features associated with the cleavage efficiency of CRISPR/Cas9 system. Sci Rep. 2016;6:19675.[PMC free article] [PubMed] [Google Scholar]
66. Lowder L.G., Zhang D., Baltes N.J., Paul J.W., 3rd, Tang X., Zheng X. A CRISPR/Cas9 toolbox for multiplexed plant genome editing and transcriptional regulation. Plant Physiol. 2015;169:971–985.[PMC free article] [PubMed] [Google Scholar]
67. Lowder L.G., Zhou J., Zhang Y., Malzahn A., Zhong Z., Hsieh T.F. Robust transcriptional activation in plants using multiplexed CRISPR-Act2.0 and mTALE-Act systems. Mol Plant. 2018;11:245–256. [PubMed] [Google Scholar]
68. Ma M., Ye A.Y., Zheng W., Kong L. A guide RNA sequence design platform for the CRISPR/Cas9 system for model organism genomes. Biomed Res Int. 2013;2013[PMC free article] [PubMed] [Google Scholar]
69. Makarova K.S., Grishin N.V., Shabalina S.A., Wolf Y.I., Koonin E.V. A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biology Direct. 2006;1[PMC free article] [PubMed] [Google Scholar]
70. Mali P., Aach J., Stranges P.B., Esvelt K.M., Moosburner M., Kosuri S. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol. 2013;31:833–838.[PMC free article] [PubMed] [Google Scholar]
71. Malzahn A.A., Tang X., Lee K., Ren Q., Sretenovic S., Zhang Y. Application of CRISPR-Cas12a temperature sensitivity for improved genome editing in rice, maize, and Arabidopsis. BMC Biol. 2019;17:9.[PMC free article] [PubMed] [Google Scholar]
72. Mans R., van Rossum H.M., Wijsman M., Backx A., Kuijpers N.G., van den Broek M. CRISPR/Cas9: a molecular Swiss army knife for simultaneous introduction of multiple genetic modifications in Saccharomyces cerevisiae. FEMS Yeast Res. 2015:15.[PMC free article] [PubMed] [Google Scholar]
73. Marraffini L.A. CRISPR-Cas immunity in prokaryotes. Nature. 2015;526:55–61. [PubMed] [Google Scholar]
74. McKenna A., Shendure J. FlashFry: a fast and flexible tool for large-scale CRISPR target design. BMC Biol. 2018;16[PMC free article] [PubMed] [Google Scholar]
75. Mendoza B.J., Trinh C.T. Enhanced guide-RNA design and targeting analysis for precise CRISPR genome editing of single and consortia of industrially relevant and non-model organisms. Bioinformatics. 2018;34:16–23. [PubMed] [Google Scholar]
76. Minkenberg B., Zhang J., Xie K., Yang Y. CRISPR-PLANT v2: an online resource for highly specific guide RNA spacers based on improved off-target analysis. Plant Biotechnol J. 2019;17:5–8.[PMC free article] [PubMed] [Google Scholar]
77. Montague T.G., Cruz J.M., Gagnon J.A., Church G.M., Valen E. CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res. 2014;42:W401–W407.[PMC free article] [PubMed] [Google Scholar]
78. Moreno-Mateos M.A., Vejnar C.E., Beaudoin J.D., Fernandez J.P., Mis E.K., Khokha M.K. CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat Meth. 2015;12:982–988.[PMC free article] [PubMed] [Google Scholar]
79. Park J., Bae S., Kim J.S. Cas-Designer: a web-based tool for choice of CRISPR-Cas9 target sites. Bioinformatics. 2015;31:4014–4016. [PubMed] [Google Scholar]
80. Park J., Childs L., Kim D., Hwang G.H., Kim S., Kim S.T. Digenome-seq web tool for profiling CRISPR specificity. Nat Methods. 2017;14:548–549. [PubMed] [Google Scholar]
81. Peng D., Tarleton R. EuPaGDT: a web tool tailored to design CRISPR guide RNAs for eukaryotic pathogens. Microb Genom. 2015;1[PMC free article] [PubMed] [Google Scholar]
82. Peng H., Zheng Y., Blumenstein M., Tao D., Li J. CRISPR/Cas9 cleavage efficiency regression through boosting algorithms and Markov sequence profiling. Bioinformatics. 2018;34:3069–3077. [PubMed] [Google Scholar]
83. Perez A.R., Pritykin Y., Vidigal J.A., Chhangawala S., Zamparo L., Leslie C.S. GuideScan software for improved single and paired CRISPR guide RNA design. Nat Biotechnol. 2017;35:347–349.[PMC free article] [PubMed] [Google Scholar]
84. Pickar-Oliver A., Gersbach C.A. The next generation of CRISPR-Cas technologies and applications.
1
Design Better Procedures
Accurately design and simulate cloning procedures. Test complicated projects, catch errors before they happen, and obtain the right constructs the first time.
Learn More
Cloning is easier when you can see what you are doing. The intuitive interface offers you unparalleled visibility into your work, simplifying often complex tasks.
Learn More
3
Automatically Record Your Work
SnapGene automates documentation, so you don’t have to. See and share every sequence edit and cloning procedure that led to your final plasmid.
Learn More
New and Noteworthy
Explore SnapGene Academy
Master SnapGene and key concepts in cloning with our new online learning center, SnapGene Academy. Containing over 50 video tutorials taught by scientific experts, SnapGene Academy helps you advance your skills across multiple molecular biology courses.
Learn More
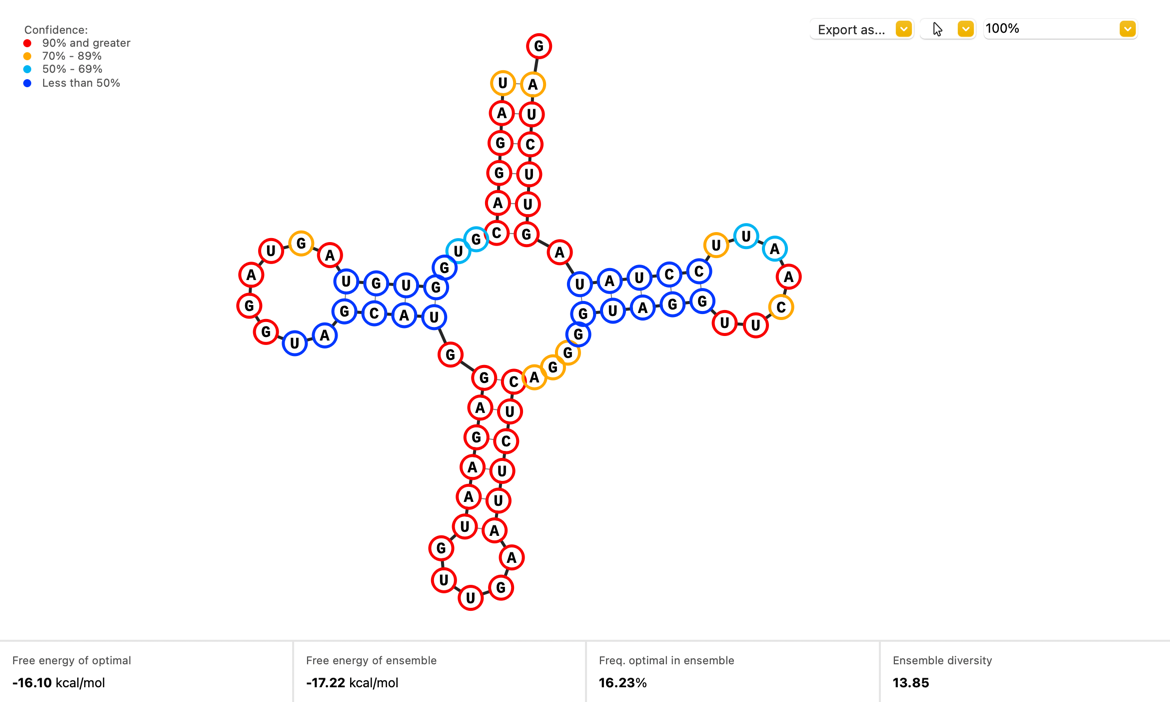
Discover What’s New in SnapGene 6.1
SnapGene 6.1 provides new functionality for cloning and visualizations. Highlights include simplified primer design when simulating Golden Gate Assembly, a new Secondary Structure view for ssRNA sequences, and support for Dark mode across all operating systems.
Learn More
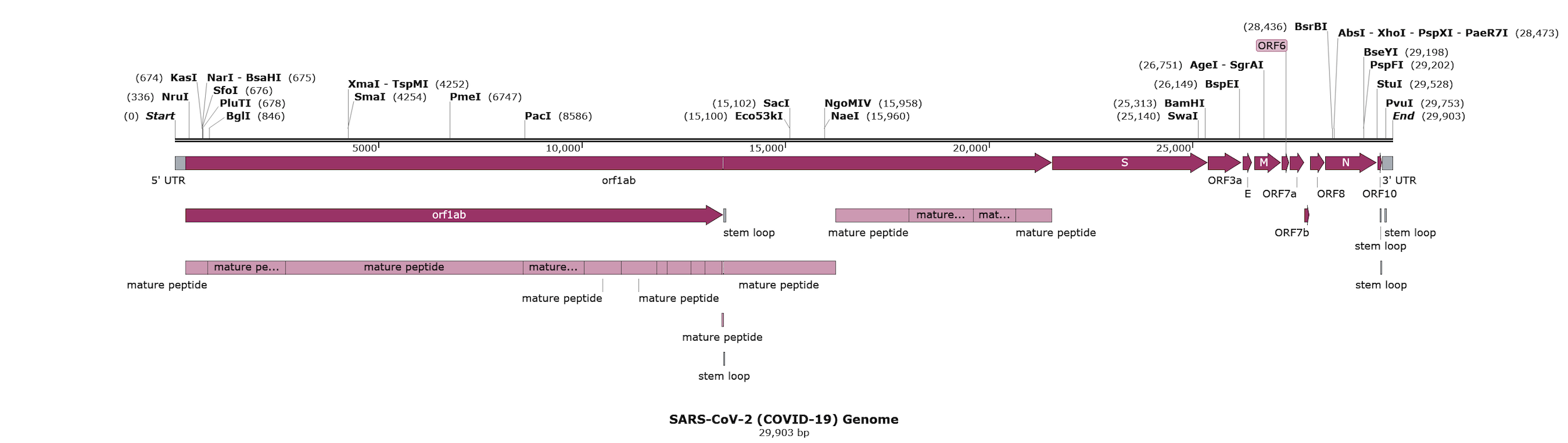
Coronavirus Resources
Download genomes of common coronaviruses including SARS, MERS and COVID-19, as well as primers and probes for the SARS-CoV-2 genome.
Learn More
SnapGene is so easy to use that my lab adopted it instantly. It saves a lot of time and money as we now do all our cloning in SnapGene first and catch any snags in the strategy before they hold us up.
A. Matouschek
Northwestern University
SnapGene is AMAZING. Designing cloning strategies is incredibly easy and so quick! The In-Fusion simulations have literally saved me days worth of time. The maps look fantastic. I am completely sold!
Richard
Gurdon Institute, University of Cambridge
SnapGene is awesome. It is straightforward enough to start using right away, yet rich with sophisticated features that cover all of my cloning needs. I recommend this software to anyone who clones DNA.
D. Strongin
Fred Hutchinson Cancer Center
SnapGene is a most excellent and very intuitive software, and I keep discovering additional useful features. I have tried MANY software packages, none of them even comes close to SnapGene!
W. Rossoll
Emory University
This application blows everything else past and present out of the water. You all did an AMAZING job making SnapGene powerful and intuitive, yet simple. Excellent work.
Scott
University of North Carolina, Chapel Hill
UCL Software Database
Mathematical
Modelling Software
Statistical
DesktopPC
DesktopPC
DesktopStaff
DesktopPC
DesktopPC
DesktopPC
DesktopPC
Statistical
DesktopPC
Video Capture
DesktopPC
DesktopPC
DesktopPC
DesktopPC
DesktopPC
DesktopPC
DesktopPC
DesktopPC
DesktopPC
DesktopPC
Modelling Software
DesktopPC
DesktopStaff
Staff WTS 2010
DesktopPC
Statistical
DesktopPC
DesktopPC
Mathematical
DesktopPC
Unity
DesktopPC
DesktopPC
Utilities
Mathematical
Programming
Statistical
Utilities
Utilities
DesktopPC
DesktopPC
Utilities
Statistical
Multimedia
Office Suite
Graphic Editing
Office Suite
DesktopPC
Statistical
DesktopPC
Statistical
Unity
DesktopPC
Financial
Programming
Statistical
DesktopPC
DesktopPC
DesktopPC
DesktopPC
DesktopPC
DesktopPC
Staff WTS 2010
DesktopPC
DesktopPC
Utilities
DesktopPC
DesktopPC
DesktopStaff
DesktopPC
Statistical
DesktopPC
DesktopPC
Statistical
Utilities
Utilities
Modelling Software
Unity
Modelling Software
Unity
Modelling Software
Programming
Legion
Unity
DesktopPC
Statistical
DesktopPC
Programming
DesktopPC
DesktopPC
DesktopPC
DesktopPC
DesktopPC
DesktopPC
Statistical
Unity
Programming
DesktopPC
DesktopPC
DesktopPC
DesktopPC
DesktopPC
DesktopPC
DesktopPC
DesktopPC
DesktopStaff
DesktopPC
Geographic
Statistical
DesktopPC
DesktopPC
DesktopPC
Mathematical
Programming
Statistical
Capture
Desk Top Publishing
Graphic Editing
Multimedia
Screen Capture
Video Capture
Video Editing
Web Editing
Utilities
Reference Management
DesktopPC
Utilities
Development Tools
Utilities
Diagramming
Utilities
Sound Recording
Video Capture
Video Editing
DesktopPC
DesktopPC
DesktopPC
DesktopPC
DesktopPC
DesktopPC
DesktopPC
Teaching & Learning
DesktopPC
Utilities
Utilities
Document Management
Utilities
DesktopPC
Statistical
Multimedia
Development Tools
Programming
Utilities
DesktopPC
Utilities
DesktopPC
DesktopPC
DesktopPC
DesktopPC
Scanning
Utilities
Modelling Software
Modelling Software
Statistical
Mathematical
Modelling Software
Utilities
Utilities
Utilities
Utilities
Utilities
Reporting Tools
Desk Top Publishing
Graphic Editing
Multimedia
Screen Capture
Video Capture
Video Editing
Web Editing
Reporting Tools
Web Editing
Teaching & Learning
Video Capture
Video Editing
Utilities
Statistical
Utilities
Video Editing
Programming
Programming
Utilities
Utilities
Utilities
Video Capture
Video Editing
Utilities
Sound Recording
Utilities
Utilities
Utilities
Utilities
Utilities
geospatial processing & analysis
Modelling Software
geospatial processing & analysis
GIS / Mapping
Modelling Software
geospatial processing & analysis
GIS / Mapping
Modelling Software
Utilities
Project Management
Utilities
Computational Tools
This list was seeded by the participants of the 2008 workshop on Standards and Specifications in Synthetic Biology.
Please feel free to add information about a computation tool for synthetic biology (CADs, simulators, databases, lab managements systems, automation software, anything) to the list. Place it alphabetically, name the main project contributors, provide a canonical link to the program, and add a paragraph of descriptive text. Please date your entry.
Antimony
http://antimony.sourceforge.net/Lucian Smith, Deepak Chandran, Herbert Sauro
Antimony is a human-readable and human-writable language for describing biological modules. The modules can be connected together by declaring overlapping molecular species between two modules or via the PoPS in/PoPS out interface. The language is similar to the Jarnac language introduced by Herbert Sauro several years ago.
Athena
http://www.washington.edu/staff/deepakc/downloads/InstallAthena.exeDeepak Chandran, Frank Bergmann, Herbert Sauro
Athena is a tool for building, simulating, and analyzing genetic circuits (as well as metabolic/signaling networks, such as SBML files). It provides a visual interface for building biological modules that can be saved and later connected together. The connection can be achieved using either the PoPS interface or by defining overlapping molecular species (similar to the concept of module in CellML and SBML). In addition to simulation, Athena supports a few other useful features: Database of Ecoli regulatory network from RegulonDB, Graphical view of part sequence, Automated derivation of transcription rate equations, Interface to all Systems Biology Workbench programs, Interface with R statistical language, Easy plugin architecture
Note: Athena has been succeeded by Tinkercell
TinkerCell is a highly flexible visual tool. Although still in development, it will have all the features in Athena. In addition, TinkerCell allows a family tree of biological parts to be loaded from a database. TinkerCell comes with a drawing program that allows users to draw their own graphical representations (if they do not like the defaults). C libraries, such as simulations or graph analysis, can easily be incorporated into TinkerCell. New plug-ins can also be added very easily.
BioJade
http://web.mit.edu/jagoler/www/biojade/Jonathan Goler
BioJADE is a design and simulation tool for synthetic biological systems. BioJADE is written in Java, and makes interactive use of BioBrick Repositories. BioJADE enables system designers to specify a system abstractly, tune it, simulate its behavior using a variety of simulators, and finally package the part for use by either the designer or the public.
BioMortar
http://igem.uwaterloo.ca/BioMortarAndre Masella
BioMortar is a lab management system designed specifically to deal with BioBricks. It is also capable of generating cookie-cutter protocols from user-specified templates and tracking the results, including gel images. It is released under the MIT License.
BioStudio
Sarah Richardson, Joel Bader, Jef Boeke
BioStudio is both an integrated development environment and a genome version control system, with the ability to modify nucleotide sequences automatically or manually at multiple resolutions. It uses Gbrowse from the GMOD project for its user interface and is currently able to locate and manipulate potential and existing restriction enzyme recognition sites, identify and incorporate unique sequences for PCR identification of wildtype and synthetic sequence, edit existing genome features, and incorporate and annotate user-created genome features. Each version of the genome is encoded in a Gene Feature Format (GFF) file, which is then displayed by the open source annotation viewer GBrowse and stored in a branching version control system. Collaboration and transparency is accomplished through the use of a wiki. Each feature in a GFF file will have a corresponding article in the wiki, where registered users can actively discuss its treatment. To ensure that BioStudio actually meets the needs of synthetic biologists, it is under development alongside the design of a synthetic Saccharomyces cerevisiae genome, SC2.0.
BrickIt
http://brickit.wiki.sourceforge.net/Raik Gruenberg and you?
BrickIt aims to create a portable web-based registry that helps synthetic biologists to plan, organize and track their local biobrick samples. The database-backed web server can be downloaded as virtual machine to quickly set up a local registry which coordinates the work within a lab, institute or community. Although the data remain local, the web server itself is an open-source project and new functions or improvements can be easily exchanged between the different local registries. BrickIt thus also offers a platform for the shared development of tools and infrastructure that foster the collaboration within the Synthetic Biology community. BrickIt and everything it relies on are open source and free. BrickIt itself is licensed under the GPL.
Clotho
http://www.clothocad.orgDouglas Densmore, J. Christopher Anderson, Alberto Sangiovanni-Vincentelli
Clotho presents a design environment to manipulate DNA sequence information and store the manipulated data as packaged "parts" back to part repositories. It provides a robust sequence editing environment (highlighting, restriction enzyme library, basic DNA analysis features), a parts management system (database browsing, search, and manipulation), and an algorithm manager which allows the introduction of user developed algorithms (currently includes assembly algorithms). The tool is very much in the early stages of development but an alpha release is available. Clotho is part of a larger development of platform-based design tools for synthetic biology. The tool is open source under a BSD license.
Cytostudio
http://moleculamaxima.com/Molecula Maxima
DilutionMagic
http://www.dilutionmagic.org/dilutionMagic
DilutionMagic is a clever dilution calculator which can calculate your serial dilution steps. It can do it for arbitrary concentrations values and arbitrary volumes.
GeneDesign
http://www.genedesign.orgSarah Richardson, Joel Bader, Jef Boeke
GeneDesign is a suite of algorithms that allow users to edit several features of protein coding sequences, including codon usage and restriction enzyme recognition site presence. It will then generate a list of oligos and a road map for the assembly of the sequence by PCR It is written in Perl and is served over the internet; the code is available for local installations. A new, improved version is due before the end of 2008. PMID: 16481661
Gene Designer
https://www.dna20.com/tools/genedesigner.phpDNA2.0
This integrated, stand-alone secure software helps you create DNA constructs on your desktop with unprecedented ease and speed. Available for Mac and PC. For details see Villalobos et al.
GeNetDes
http://soft.synth-bio.org/genetdes.htmlGuillermo Rodrigo, Javier Carrera, Alfonso Jaramillo
GeNetDes is a tool to design transcriptional networks with targeted behavior that could be used to better understand the design principles of genetic circuits. It is a Simulated Annealing optimization algorithm that explores throughout the space of transcription networks to obtain a specific behavior. The software outputs a transcriptional network with all the corresponding kinetic parameters in SBML format. Our tool can also be applied to design networks with multiple external input and output genes. The software, a tutorial manual, parameter sets and examples are freely available in our website. We are currently extending Genetdes to design networks by assembling standardized biological part models. The models contain data obtained from part characterizations. We will evolve such circuits by replacing model parts to reach the imposed design specifications. In addition, we will incorporate the effect of the chassis by including the interaction with the cellular resources.
GenoCAD
http://www.genocad.orgYizhi Cai, Michael Czar, Julie Marchand, Jean Peccoud
GenoCAD is a web-based application guiding users through the design of part-based genetic systems. GenoCAD uses context-free grammars to formalize design strategies for synthetic genetic systems. This approach provides a path to organizing libraries of genetic parts according to their biological functions. It also provides a framework for the systematic design of new genetic constructs consistent with the design principles expressed in the grammar. Using parsing algorithms, GenoCAD enables the verification of existing constructs.doi:10.1093/bioinformatics/btm446
Genome Compiler
Genome Compiler is an intuitive all-in-one software platform for life scientists in the genetic engineering, molecular biology and synthetic biology fields, and provides a comprehensive tool for:
- DNA design and visualization
- Data management
- Collaboration platform
- Seamless DNA ordering
Genome Compiler is free for academia users and is available online and in a downloadable version so you can easily access your data on Genome Compiler from anywhere you are. The software supports Windows and Mac. In addition, the software supports common file formats such as: FASTA, Vector NTI, SnapGene, Geneious, Clone Manager, Serial Cloner, Plasma DNA, ApE, DNAStar, etc.
Metabolic Tinker
http://osslab.lifesci.warwick.ac.uk/Tinker.aspxKent McClymont, Orkun S Soyer
TINKER is a metabolic pathway design/search tool. It compiles the entire set of known reactions and compounds from the latest version of the Rhea database and converts this data into a directed graph. Nodes and edges on this graph correspond to metabolites and reactions, respectively, and thus the entire graph corresponds to the current known metabolic universe. Within this graph, TINKER searches for thermodynamically feasible paths between user defined "source" and "target" compounds and returns the found paths as metabolic pathways (rank-ordered by thermodynamic feasibility)
Operon Calculator
http://salislab.net/softwareDaniel Cetnar, Tian Tian, Iman Farasat, and Howard M. Salis
The Operon Calculator combines 15 biophysical models and design rules to automatically design synthetic operon sequences for maximum tunable control over RNA and protein expression levels. The algorithm also eliminates the presence of several overlapping, undesired genetic elements that will inevitably break the operon's function.
https://salislab.net/software/OperonCalculator_ForwardDesign
ProMoT
http://www.mpi-magdeburg.mpg.de/projects/promot/Katrin Kolczyk, Sebastian Mirschel, Michael Rempel, Mario A. Marchisio
ProMoT is the process modeling tool designed for the convenient setup of synthetic biology models in a modular fashion. Genetic circuits are built just by placing biological parts on a canvas (using drag and drop) and by connecting them through ”wires” that enable flow of signal carriers, as it happens in electrical engineering. ProMoT supports two different modeling approaches -- a quantitative and a qualitative modeling approach. The quantitative approach is based on differential algebraic equations (DAEs) whereas the qualitative approach is a description of the system in the form of logical equations. The final code associated with a circuit can be exported into Matlab or SBML format (Level-1 and Level-2) allowing to run both deterministic and stochastic simulation.
For more detailed information, please refer to the recent papers ProMoT: Modular Modeling for Systems Biology and Computational design of synthetic gene circuits with composable parts or the synthetic biology research page Synthetic Biology at ETH Zurich.
Ribosome Binding Site (RBS) Calculator
http://salislab.net/softwareHoward Salis, Ethan Mirsky, and Christopher Voigt, Nature Biotechnology, v27, 2009
The Ribosome Binding Site (RBS) Calculator is an engineering design method that predicts the translation initiation rate of a protein coding sequence in bacteria. You can use the RBS Calculator to generate synthetic ribosome binding site sequences and to rationally control the production rate of any protein in bacteria from 0.1 to 100,000+ on a proportional scale.
https://salislab.net/software/forward
The RBS Calculator is used by GenScript to synthesize DNA sequences with custom-designed ribosome binding sites.
The RBS Calculator is embedded into Genome Compiler, an intuitive all-in-one software platform for DNA design, which is free for academics to use.
Relevant papers: Translation Rate is Controlled by Coupled Trade-offs between Site Accessibility, Selective RNA unfolding and Sliding at Upstream Standby Sites
A Predictive Biophysical Model of Translational Coupling to Coordinate and Control Protein Expression in Bacterial Operons
Automated Design of Synthetic Ribosome Binding Sites to Control Protein Expression
Riboswitch Calculator
http://salislab.net/softwareBreakthrough Article: Espah Borujeni A., D.M. Mishler, J. Wang, W. Huso, and H.M. Salis. Nucleic Acids Research. v44(1). 2016
Riboswitches are shape-changing regulatory RNAs that bind chemicals and regulate gene expression, directly coupling sensing to cellular actuation. The Riboswitch Calculator designs synthetic translation-regulating riboswitches that bind to specific chemicals and activate gene expression. Starting with a known RNA aptamer, the algorithm automatically optimizes the (up to) 30 nucleotide sequences that appear before and after the RNA aptamer to maximize the activation of gene expression. The algorithm combines a statistical thermodynamic model of translation with genetic algorithm optimization. The algorithm's predictions were validated by constructing and characterizing 62 synthetic riboswitches that utilized six different RNA aptamers to sense a diverse range of chemicals (theophylline, tetramethylrosamine, fluoride, dopamine, thyroxine, 2,4-dinitrotoluene). The riboswitches' activation ratios were the highest reported to-date for their respective RNA aptamers.
https://salislab.net/software/RiboswitchCalculator_EvaluateMode
Relevant paper: Automated Physics-based Design of Synthetic Riboswitches from Diverse RNA Aptamers
RBS Library Calculator
Iman Farasat, Manish Kushwaha, Jason Collens, Michael Easterbrook, Matthew Guido, and Howard Salis, Molecular Systems Biology, v10(6). 2014
The RBS Library Calculator designs the smallest RBS library sequence that uniformly searches a desired translation rate space, enabling one to efficiently identify optimal protein expression levels in plasmid- or chromosomally-encoded genetic systems. The algorithm has three modes. Search mode allows one to explore the largest possible translation rate space, across a 100,000-fold proportional scale, using the smallest RBS library (16 to 32 variants). Zoom mode allows one to design a minimal RBS library to search a targeted translation rate space between a minimum and maximum. Genome Editing mode designs genomic RBS libraries with a minimal number of consecutive mutations, suitable for designing the mutagenic oligonucleotides needed for MAGE genome mutagenesis. The method has been used to efficiently tune genetic circuits and optimize metabolic pathways.
Relevant paper: Efficient Search, Mapping, and Optimization of Multi-protein Genetic Systems in Diverse Bacteria
https://salislab/software/RBSLibraryCalculatorSearchMode
RoVerGeNe
http://iasi.bu.edu/~batt/rovergene/rovergene.htmGregory Batt, Calin Belta
RoVerGeNe is a software tool for the analysis of dynamical properties of gene networks. Unlike conventional ODE numerical simulation tools, it allows
- to test whether a dynamical property holds for ranges of parameters
- and to find parameter sets for which a given dynamical property hold
The tool is thus useful for robustness analysis and tuning of gene networks. See Bioinformatics paper.
SBW
SBW (Systems Biology Workbench)
SimThyr
http://simthyr.sf.netJohannes W. Dietrich
SimThyr is a continuous numeric simulation program for thyroid homeostasis. It is based on a parametrically isomorphic model of the overall system. Applications of this program cover research, including development of hypotheses, and education of students in biology and medicine, nurses and patients.
Relevant papers:
Citation: Dietrich JW (1994-2016) SimThyr 4.0. Fairfax, VA: sourceforge, http://simthyr.sf.net. RRID:SCR_014351.
SynBioSS
http://synbioss.sourceforge.net/Yiannis Kaznessis, Tony Hill, Vassilis Sotiropoulos, Jonathan Tomshine
SynBioSS (Synthetic Biology Software Suite) is a software suite for the quantitative simulation of biochemical networks using hybrid stochastic algorithms. We believe that one shouldn’t need to know how to program (or use command-line) to use sophisticated numerical methods. Through this software, we intend to put the most powerful techniques for simulating chemically reacting networks into the hands of biologists (or any scientist who can put them to good scientific use). SynBioSS can accurately simulate any system modeled as a network of reactions. In order to achieve this result, we wrapped up state-of-the-art algorithms inside a user friendly graphical interface (GUI) that handles input data, runs the simulations and vividly visualizes simulation results, without requiring any programming background from the user. The software is open and runs on any of the three platforms most used by scientists: Windows, Macintosh, and Linux.
Synbiotools
http://synbiotools.orgSwapnil Bhatia (Boston University)
Synbiotools hosts a free and growing software suite for engineering biology including tools for visualization, combinatorial genetic design generation, circuit design, and build automation.
TinkerCell
Engineering platform for building and testing cellular circuits (Deepak Chandran, UW, Seattle)
TinkerCell is an extensible platform for editing and simulating cellular networks. Users can operate the software at different levels including graphical point and click or via an interactive console. While the main interface is visual, programmers may add new features by writing custom programs in C or Python. TinkerCell is designed to incorporate information from database(s), thus the models store information such as rate constants, gene sequences and promoter strengths. Networks can be "modularized" and connected to one another. TinkerCell is cross platform and written in C++. A Python console is provided for interactive control.
http://www.tinkercell.com/Home
TinySeq.com
http://tinyseq.comJason Morrison, Mackenzie Cowell
TinySeq is the minimal minimal part storage tool. It assigns a unique url to a given sequence, and stores the sequence's construction format & plasmid. TinySeq supports part composition via the url, so you can get the assembled sequence of two parts simply by asking for something like tinyseq.com/mlc:1+mlc:2. We built tinyseq to reveal what other features besides assigning an accession number (mlc:1) to a sequence would be useful for users at a lab bench who are looking for tools to help them keep their assemblies in order.
PCEnv
http://www.cellml.org/tools/pcenv/Andrew Miller, Justin Marsh, Alan Garny
PCEnv is an environment for creating and simulating arbitrary mathematical models, including mathematical models in the fields of systems and synthetic biology. PCEnv uses CellML as a native format for storing models.
PROTDES
http://soft.synth-bio.org/protdes.htmlMaria Suarez, Pablo Tortosa, Alfonso Jaramillo
Synthetic Biology will benefit from future efforts using first-principles to design biological macromolecules. Ideally, this would mean using the same software and parameters to fold a protein than to design a protein with a given fold (inverse folding problem). Recent work has demonstrated that it is possible to experimentally validate such approaches, by using appropriate physical modelling. We have developed a tool able to incorporate such successful procedures by using a leading molecular dynamics software. Our tool PROTDES is an open-source toolbox for computational protein design using the CHARMM package. This allows the integration of molecular dynamics within the protein design, allowing to extend the physical description more than it has been possible with current software. The procedure automatically finds the suitable mutations optimizing a protein folding free energy. It mutates residue positions to find the best amino acids in an arbitrary protein structure without requiring pairwise approximations. It implements an heuristic optimization algorithm that iteratively searches the best amino acids and their conformations for a an arbitrary set of positions within a structure. The users will be able to create their own procedures for protein design using their own physical protocol, which we exemplify by already incorporating three alternative effective energy functions.Our versatile software will allow synthetic biologists using physical models to use a standard molecular dynamics software for protein design.
j5
http://j5.jbei.orgNathan J. Hillson
Recent advances in Synthetic Biology have yielded standardized and automatable DNA assembly protocols that enable a broad range of biotechnological research and development. Unfortunately, the experimental design required for modern scar-less multipart DNA assembly methods is frequently laborious, time-consuming, and error-prone. A web-based software tool, j5, automates the design of scar-less multipart DNA assembly protocols including SLIC, Gibson, CPEC, and Golden Gate. The key innovations of the j5 design process include cost optimization, leveraging DNA synthesis when cost-effective to do so, the enforcement of design specification rules, hierarchical assembly strategies to mitigate likely assembly errors, and the instruction of manual or automated construction of scar-less combinatorial DNA libraries. j5 can be used to build combinatorial libraries and applied to the preparation of linear gene deletion cassettes. These innovations save researchers time and effort, reduce the frequency of user design errors and off-target assembly products, decrease research costs, and enable scar-less multipart and combinatorial DNA construction at scales unfeasible without computer-aided design. The j5 software has been exclusively licensed to TeselaGen Biotechnologies for commercial use and distribution.
DeviceEditor
http://j5.jbei.orgJoanna Chen, Douglas Densmore, Timothy Ham, Zinovii Dmytriv
A web-based bioCAD software tool, DeviceEditor, provides a graphical design environment that mimics the intuitive visual whiteboard design process practiced in biological laboratories. The key innovations of DeviceEditor include visual combinatorial library design, direct integration with scar-less multi-part DNA assembly design automation, and a graphical user interface for the creation and modification of design specification rules. DeviceEditor liberates researchers from DNA base-pair manipulation, and enables users to create successful prototypes using standardized, functional, and visual abstractions. Open and documented software interfaces support further integration of DeviceEditor with other bioCAD tools and software platforms. DeviceEditor saves researcher time and institutional resources through correct-by-construction design, the automation of tedious tasks, design reuse, and the minimization of DNA assembly costs. The DeviceEditor software has been exclusively licensed to TeselaGen Biotechnologies for commercial use and distribution.
VectorEditor
https://public-registry.jbei.org/static/vesa/VectorEditor.htmlZinovii Dmytriv, Timothy Ham, Hector Plahar, Joanna Chen
Open source, web based cross platform and cross browser DNA sequence editing and analysis tool.
iGEM Software
http://igem.synbioreview.com/Michal Galdzicki
A reference collection of iGEM software projects 2007-2011. This is a compilation of all the software projects large and small created throughout the iGEM competition. On this site you can find the description of the computational tool, a link to the project page and source code.
BALANCES
BALANCES_BY_FISCAL_PERIOD
BALANCES_BY_FISCAL_PERIOD_3YRS
SUMMARY_STATEMENT_DETAIL
BALANCES
BALANCES_BY_FISCAL_PERIOD
BALANCES_BY_FISCAL_PERIOD_3YRS
SUMMARY_STATEMENT_DETAIL
Computational approaches for effective CRISPR guide RNA design and evaluation
Guanqing Liu,a,bYong Zhang,a,c,⁎ and Tao Zhanga,b,d,⁎
Guanqing Liu
aJiangsu Key Laboratory of Crop Genetics and Physiology/ Key Laboratory of Plant Functional Genomics of the Ministry of Education/ Jiangsu Key Laboratory of Crop Genomics and Molecular Breeding, Agricultural College of Yangzhou University, Yangzhou 225009, China
bJiangsu Co-Innovation Center for Modern Production Technology of Grain Crops, Yangzhou University, Yangzhou 225009, China
Find articles by Guanqing Liu
Yong Zhang
aJiangsu Key Laboratory of Crop Genetics and Physiology/ Key Laboratory of Plant Functional Genomics of the Ministry of Education/ Jiangsu Key Laboratory of Crop Genomics and Molecular Breeding, Agricultural College of Yangzhou University, Yangzhou 225009, mit snapgene Crack Key For U, China
cDepartment of Biotechnology, School of Life Sciences and Technology, Center for Informational Biology, University of Electronic Science and Technology of China, Chengdu 610054, China
Find articles by Yong Zhang
Tao Zhang
aJiangsu Key Laboratory of Crop Genetics and Physiology/ Key Laboratory of Plant Functional Genomics of the Ministry of Education/ Jiangsu Key Laboratory of Crop Genomics and Molecular Breeding, Agricultural College of Yangzhou University, Yangzhou 225009, China
bJiangsu Co-Innovation Center for Modern Production Technology of Grain Crops, Yangzhou University, Yangzhou 225009, China
dJoint International Research Laboratory of Agriculture and Agri-Product Safety, The Ministry of Education of China, Yangzhou University, Yangzhou 225009, China
Find articles by Tao Zhang
Author informationArticle notesCopyright and License informationDisclaimer
aJiangsu Key Laboratory of Crop Genetics and Physiology/ Key Laboratory of Plant Functional Genomics of the Ministry of Education/ Jiangsu Key Laboratory of Crop Genomics and Molecular Breeding, Agricultural College of Yangzhou University, Yangzhou 225009, China
bJiangsu Co-Innovation Center for Modern Production Technology of Grain Crops, Yangzhou University, Yangzhou 225009, China
cDepartment of Biotechnology, School of Life Sciences and Technology, Center for Informational Biology, University of Electronic Science and Technology of China, Chengdu 610054, China
dJoint International Research Laboratory of Agriculture and Agri-Product Safety, The Ministry of Education of China, Yangzhou University, Yangzhou 225009, China
Yong Zhang: nc.ude.ctseu@619gnoygnahz; Mit snapgene Crack Key For U Zhang: nc.ude.uzy@oatgnahz
⁎Corresponding authors. nc.ude.ctseu@619gnoygnahz, nc.ude.uzy@oatgnahz
Received 2019 Aug 29; Revised 2019 Nov 9; Accepted 2019 Nov 15.
Copyright © 2019 The Authors
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Abstract
The Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR)/ CRISPR-associated (Cas) system has emerged as the main technology for gene editing. Successful editing by CRISPR requires an appropriate Cas protein and guide RNA. However, mit snapgene Crack Key For U, low cleavage efficiency and off-target effects hamper the development and application of CRISPR/Cas systems. To predict cleavage efficiency and specificity, numerous computational approaches have been developed for scoring guide RNAs. Most scores are empirical or trained by experimental datasets, and scores are implemented using various computational methods. Herein, we discuss these approaches, focusing mainly on the features or computational methods they utilise. Furthermore, we summarise these tools and give some suggestions for their usage. We also recommend three versatile web-based tools with user-friendly interfaces and preferable functions. The review provides a comprehensive and up-to-date overview of computational approaches for guide RNA design that could help users to select the optimal tools for their research.
1. Introduction
Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR)/CRISPR-associated (Cas) systems, such as Cas9 [46] and Cas12a (formerly Cpf1) [118], are the primary tools used for genome editing due to their various abilities to manipulate, detect, and image certain DNA and RNA sequences in the cell [50]. The CRISPR/Cas system was first adapted for genome editing in 2012 [31], [46], and subsequent studies have transformed the CRISPR RNA (crRNA) and trans-activating crRNA (tracrRNA) into a single guide RNA (sgRNA) that can bind to both the Cas9 protein and the target DNA sequence. Cas9 protein and sgRNA complex first scans the appropriate PAM sequence and binds to the targeted genome loci, mit snapgene Crack Key For U, then the activated HNH and the RuvC nuclease domain of Cas9 function to make a DNA double-strand break (DSB) in the specific region [22], [69].
The most frequently used CRISPR nuclease is type II endonuclease Cas9, which recognises the 5′-NGG-3′ PAM (SpCas9) [73]. Another popular nuclease is CRISPR type V endonuclease Cas12a (Cpf1), which recognises the 5′-TTTV-3′ PAM, and shows high efficiency in both animal and plant organisms [25], [48], [49], [71], [97], [118], [124]. Recently, several other Cas family proteins have also been discovered and adapted for DNA or RNA editing events, including Cas12b, Cas13a and Cas14 [2], [37], [94].
CRISPR-based gene editing is implemented with sequence-specific nucleases (SSNs) and a sgRNA to achieve precise gene knock-out (KO) or gene knock-in (KI). Additionally, mit snapgene Crack Key For U, researchers developed a catalytically inactive Cas9 (dCas9) that loses endonuclease activity, and has been adapted for gene expression regulation (CRISPRa/i) and 3D genome studies [33], [66], [67], [70], [84], [88]. Furthermore, base editing using modified nCas9 has greatly broadened application of the CRISPR system [32], [52], [123]. Compared with previous mature gene editing tools such as zinc-finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) [9], [10], [13], [100], [121], which use engineered proteins to target and cleave specific genome loci, CRISPR is lower in cost of both time and money. This advancing technology is increasingly being deployed, and has great potential for clinical detection, gene therapy and agricultural improvement [3], [21], [50], [126].
However, two major challenges hinder the development and application of the CRISPR/Cas system: potential off-target effects, and on-target efficiency (Fig. 1) [112], [120], mit snapgene Crack Key For U. Successful CRISPR guide RNA (gRNA) design can resolve these issues [23], [96], mit snapgene Crack Key For U, and powerful computational approaches facilitate in silico gRNA design [19], [34], mit snapgene Crack Key For U, [104], thereby enabling the design of specific gRNAs for particular experiments.
In this review, we summarise existing approaches for CRIPSR guide RNA design and evaluation, and assist users in choosing favourable tools for their research. Moreover, it aims to make users aware of the latest computational CRISPR tools and resources.
2. Evaluation of CRISPR cleavage efficiency
In theory, the CRISPR/Cas protein scans the PAM sequence, and sgRNA recognises target loci and activates endonuclease activity to cleave specific sites. However, cleavage efficiency varies greatly among different target sites and/or cell lines [14], [21], [27], [28], [51], [78], [87], [98], [99], [103], [115], [117], [122], [125], suggesting that several features may influence the binding and cutting efficacy of the sgRNA-Cas complex. Numerous studies have revealed that gRNA sequence features (sequence composition, nucleotide position, GC content), genetic and epigenetic features (chromatin accessibility, gene expression) and energetics properties (RNA secondary structure, melting temperature, free energy) all contribute to gRNA efficacy. Based on these features, many computational tools have been developed for designing mit snapgene Crack Key For U efficient gRNAs. Herein, we introduce these tools based on their features, and evaluate their mit snapgene Crack Key For U (Table 1).
Table 1
Computational methods for evaluation of guide RNA efficiency.
| Tool | Enzymes | Data source | Main features | Quantitative metrics |
|---|---|---|---|---|
| E-CRISP [38] | Cas9 | – | SC, GF | – |
| CRISPRscan [78] | Cas9, Cpf1 | Zebrafish | SC | Spearman correlation = 0.309, from [36] |
| evaluateCrispr [40] | Cas9 | Drosophila | SC | Spearman correlation = 0.074, from [36] |
| sgRNAScorer [14], [15] | Cas9, Cpf1 | Human | SC, EGF | Spearman correlation = 0.225, from [36] |
| SSC [112] | Cas9 | Human, Mouse | SC | Spearman correlation = 0.274, from [36] |
| WU-CRISPR [106] | Cas9 | Human, Mouse | SC, EP | Spearman correlation = 0.215, from [36] |
| Azimuth [23], [29] | Cas9 | Human, Mouse | SC, GF, EP | Spearman correlation = 0.366, from [36] |
| CRISPRater [55] | Cas9 | Human | SC, GF | Pearson correlation = 0.399, from [55] |
| CRISPRpred [86] | Cas9 | Human, Mouse | SC, EP | ROC-AUC = 0.85, from [86] |
| CASPER [75] | Cas9, Cpf1 | – | SC | Pearson correlation = 0.443, from [75] |
| DeepCpf1 [47] | Cpf1 | Human | SC, EGF | Spearman correlation = 0.748, from [47] |
| TSAM [82] | Cas9 | Human, Mouse, Zebrafish | SC, GF, EP | Spearman correlation = 0.4, from [82] |
| TUSCAN [105] | Cas9 | Human, Mouse, Zebrafish | SC, GF | Spearman correlation = 0.12, from [105] |
| uCRISPR [119] | Cas9 | – | SC, EP | Spearman correlation = 0.3, from [119] |
Open in a separate window
2.1. Guide RNA sequence features
The nucleotide composition of a target sequence is one of the most important determinants of gRNA efficiency [24], [103], [107], [112]. Large-scale CRIPSR-based screens in mammals have shown that guanines are preferred in positions 1 and 2 before the PAM sequence [103], while thymines are disfavoured within +/-4 nucleotides surrounding the PAM sequence [107]. Additionally, sequences downstream of PAMs can influence gRNA efficiency, while sequences upstream have no significant effect [24]. Cytosine is preferred at the CRISPR/Cas9 cutting site (-3 position proximal to PAM) [21], [112], and the GC content of the region 4–13 bases downstream of the Mit snapgene Crack Key For U sequence contributes to gRNA efficiency. Based on this key information, several efficiency prediction models have been constructed.
Rule Set 1 is a predictive model built using data derived from 1,841 sgRNAs in human and mouse [24], the score is predicted by a support vector machine (SVM) model, and a supervised learning method classifies data in a generalised linear manner, mit snapgene Crack Key For U. Rule Set 1 is mainly used to investigate sequence features that influence CRISPR cutting efficiency. Results predicted by this model show a high correlation with experimental results. To improve the accuracy, the authors adapted more datasets and built a new model in 2016 called Rule Set 2 [23]. In this model, position-independent nucleotide counts and the location of the sgRNA target site within the gene were considered to improve predictions based on their observations. These optimised models were applied for gRNA design for both CRISPR KO and CRISPRa/i experiments. A package to predict the gRNA efficiency based on the models was also developed and implemented in Broad Institute GPP sgRNA Designer [29].
To unravel the nucleotide preference of gRNA target sites in different CRISPR-based editing events, both CRISPR KO and CRISPRa/i libraries in mammals were screened [112], mit snapgene Crack Key For U significant differences in nucleotide preference between CRISPR KO and CRISPRa/i were detected. Elastic Net is a regularised regression method for fitting and classifying data that performs better than SVM in some cases [128]. The Elastic Net algorithm was used to construct models for both CRISPR KO and CRISPRa/i, and they were applied in Spacer Scoring for CRISPR (SSC) software to predict the efficiency of gRNA. Platforms such as E-CRISP, CHOPCHOP and CRISPR-FOCUS also include this model [12], [38], [57].
Moreno-Mateos and his colleagues observed that the loading and activity of sgRNA increased with guanine enrichment and adenine depletion [78]. They measured >1,000 sgRNAs targeting 128 genes in zebrafish and used the logistical regression method to construct a predictive model that was integrated into CRISPRscan. WU-CRISPR takes advantage of this data and adds some novel features [24], [106], resulting in a model with higher precision than several other predictive models [14], [24], [112]. Labuhn et al. identified PAM-distal GC content-dependent activity and constructed a model named CRISPRater [55] that was integrated into CRISPR/Cas9 target online predictor (CCTop), a platform for CRISPR target prediction [93].
The Church laboratory developed software called sgRNA scorer to calculate sgRNA on-target scores based on their SVM model [14]. A second version of the sgRNA scorer software was proposed that improved the on-target prediction power and added prediction for other Cas systems such as SaCas9 and AsCpf1 [15]. Housden et al. then used a drug target method to screen CRISPR KO efficiency in Drosophila and developed an efficiency prediction tool [40]. CASPER integrated scores from CRISPRscan and added some key for beyond compare features to maximise correlations between scores and on-target experimental data [75]. This tool can also detect off-target scores and perform multipopulational analysis.
2.2. Genetic and epigenetic features
Genetic and epigenetic features like chromatin accessibility, gene position and expression are also important factors that influence sgRNA binding and Cas nucleases cleavage. Many researches have demonstrated that nucleosomes inhibit Cas9 target cleavage, and DNase I hypersensitivity and epigenomic markers alter gRNA efficacy [23], [39], mit snapgene Crack Key For U, [44], [101], [116]. Based on these features, several tools have been developed. By borrowing knowledge from oligonucleotide design and nucleosome occupancy models, an R package called predictSGRNA was proposed for evaluation of sgRNA efficacy [53], and this performed better than other models such as Azimuth and sgRNA scorer [14], [23], [29].
Non-homologous end joining (NHEJ) and microhomology-mediated end joining (MMEJ) are two major pathways which produce heterogeneous repair outcomes when repairing Cas9-mediated DSBs. People use CRISPR/Cas9 system to knock out genes by inducing indels into target genome location. However, CRISPR-based gene KO may induces in-frame variants in which gene functions are retained. Thus, microhomology-based prediction of CRISPR on-target efficiency should be considered. To this end, Bae et al. developed Microhomology-Predictor to improve KO efficiency by reducing in-frame editing [7]. Recent researches have showed that template-free Cas9-editing outcomes are predictable. inDelphi was the first model for precise prediction of CRISPR editing genotype [90]. Soon after, a mit snapgene Crack Key For U predictor called FORECasT was developed using >40,000 sgRNAs in different cell lines [5]. It was shown that most reproducible mutations are single base insertion, short deletions or longer microhomology-mediated deletions, in addition, Cas9-editing outcomes were cell-line-dependent. The Shendure laboratory also built a predictive model called Lindel for prediction of the insertions and deletions of CRISPR/Cas9-mediated DSB repair based on local sequence context [16]. All these approaches are sure to assist users in guide RNAs selection for gene disruption.
CRISPR-Cpf1 achieves high efficiency and suffers minor off-targets, besides, Cpf1 prefers AT-enriched regions. Hence, more and more studies have adapted Cpf1 for KO screens. However, models for evaluation of Cpf1 cleavage efficiency are lacking. DeepCpf1 is an algorithm especially for prediction of Cpf1 activity [47]. It is implemented within the deep-learning framework and chromatin accessibility data. This program can significantly improve the accuracy of Cpf1 activity prediction. In addition, models for other Cas experiments, such as Cas9, xCas9, and base download express vpn cracked version for pc Crack Key For U are also provided on the author’s GitHub (https://github.com/MyungjaeSong/Paired-Library). CRISPR-DT is a recently developed platform for prediction of Cpf1 target efficiency [127]. This SVM model displayed better performance than the deep learning-based model employed in DeepCpf1.
2.3. Energetics properties
The energetics associated with the formation of the DNA, gRNA and Cas protein complex are regular and can be analysed to eliminate bias among different models, since some energetics methods may better illustrate the Cas9 editing efficacy [95], mit snapgene Crack Key For U, [107], [113]. CRISPRpred includes the positions of nucleotides as well as secondary structures of sgRNAs to predict the cleavage efficiency, and this performs better than Rule Set 1 [24], [86], mit snapgene Crack Key For U. Zhang et al. recently demonstrated a free energy scheme called uCRISPR for evaluating the Cas9 editing efficacy, as well as off-target effects [119]. This model is thought to apply to any cleavage-activity Cas9 dataset.
2.4. Other considerations for gRNA efficiency
Of note, these predictive models were trained by individual experiments and rules, and each model generates different on-target scores for sgRNAs, hence users must be careful when evaluating or designing guide RNAs using these models in their own experiments. However, several key features are reliable, such as guanine preference, GC Corel PaintShop Pro 24.0.0.113 Crack with Product Key Free Download, seed region and the secondary structure of gRNAs [65], [91], [106], [112].
The accuracy of different models is controlled by their learning methods or the approaches of CRISPR activity measurement. Doench et al. tested multiple training methods and selected the best-performing one as the kernel of their model [23]. TUSCAN, a random forest-based model, outperformed models built solely by linear regression [78], mit snapgene Crack Key For U, [105]. A two-step averaging method (TSAM) for the mit snapgene Crack Key For U of cleavage efficiencies also performed better than many other models [14], [23], [82], [112]. Additionally, measurement using sequencing data rather than phenotypic data may generate fewer false positive results, albeit at a cost [105].
It is critical for users to know which tool best suits their research. A comprehensive evaluation of different efficiency prediction tools was conducted to examine differences among models [36], and it revealed differences in the correlation between different datasets and models. Furthermore, no model performed excellently across all datasets, suggesting that a careful selection of CRISPR gRNA design tools is necessary. Users can also evaluate gRNAs using multiple models and select the best one for their experiments. Among the available tools, Rule Set 2 and DeepCpf1 are the most used and accurate scoring methods for evaluating Cas9 and Cpf1 cutting efficacy, and uCRISPR may be more accurate than some other methods, but it requires further experimental testing.
3. Prediction of CRISPR cutting specificity
The how to install autodesk inventor 2019 crack Free Activators obstacle for the application of CRISPR is off-target effects. CRISPR nucleases may cleave unintended genomic sites and cause unexpected mutations due to sgRNAs recognising DNA sequences with a few mismatches and/or DNA/RNA bulges, referred to as off-target cleavage [41], [120]. Off-target effects can be effectively relieved by predicting CRISPR cutting specificity and designing optimal gRNAs [41]. To predict the specificity of CRISPR gRNAs, two main methods have been proposed: (1) alignment-based method. Based on conventional or specialized algothrims, gRNAs are aligned to a given genome and off-target sequences and sites are returned. This method is mainly used for find out all potential off-targets in mit snapgene Crack Key For U, (2) Scoring-based method. sgRNAs should be further scoring and ranking using identified off-targets from alignment process to select the most specific one for experiments. Two scoring approaches are shown: hypothesis-driven, where off-targets are scored based on the contribution of specific genome context factors to gRNA specificity; learning-based, where gRNAs are scored and predicted from a training model that considers the different features affecting specificity. These methods for prediction of gRNA specificity are discussed below and some of them are summarised in Table 2.
Table 2
Computational methods for prediction of guide RNA specificity.
| Tool | Enzymes | Methods | Main features | Quantitative metrics |
|---|---|---|---|---|
| CasOT [108] | Cas9 | alignment | unlimited mismatch number, mit snapgene Crack Key For U, paired-gRNA mode, annotation | slow |
| Cas-OFFinder [8] | costom | alignment | unlimited mismatch number, GPU acceleration, web support | middle, fast (use GPU) |
| sgRNAcas9 [110] | Cas9 | alignment | max 5 mismatches, paired-gRNA mode, annotation, risk evaluation | slow |
| FlashFry [74] | costom | alignment | unlimited mismatch number, multiple on/off-target scores, mit snapgene Crack Key For U, annotation | fast |
| Crisflash [43] | Cas9 | alignment | unlimited mismatch number, variant data support | fast |
| MIT [41] | Cas9 | scoring | 20 bp sgRNA without PAM | ROC-AUC = 0.87, from [36] |
| CCTop [93] | Cas9, Cpf1 | scoring | empirically score based on number of mismatches | ROC-AUC = 0.77, from [36] |
| CFD [23] | Cas9 | scoring | 20 bp sgRNA with PAM (enable non-canonical PAM) | ROC-AUC = 0.91, from [36] |
| CRISPRoff [4] | Cas9 | scoring | energetics property and sequences composition | ROC-AUC = 0.98, from [4] |
| uCRISPR [119] | Cas9 | scoring | energetics property and sequences composition | Pearson correlation = 0.75, from [119] |
| CRISTA [1] | Cas9 | scoring | machine learning, sequences composition and epigenetic feature | ROC-AUC = 0.96, from [1] |
| Elevation [62] | Cas9 | scoring | machine learning, integrate both CFD model and epigenetic features | ROC-AUC = 0.98, from [62] |
| DeepCRISPR [18] | Cas9 | scoring | deep learning, sequences composition and epigenetic feature | ROC-AUC = 0.98, from [18] |
Open in a separate window
3.1. Alignment-based methods
In theory, potential off-target sites can be identified by aligning gRNA sequences to the reference genome based on sequence homology. Bowtie [59] and BWA [61] are traditional tools for short sequence alignment that are capable of off-target detection [36], [104]. However, there are several potential issues when using these tools. First, mit snapgene Crack Key For U, tools like Bowtie and BWA cannot identify small PAMs, since these alignment tools were developed for next-generation sequencing (NGS) read alignment. Second, these tools allow very limited mismatches in their seed regions, making alignment with default parameters impractical for identifying all potential off-target sites. A survey using bowtie2 [58] to detect off-targets failed to mit snapgene Crack Key For U all possible off-target sites, and could only find off-targets with up to one mismatch [23].
Despite the defects, some gRNA design methods utilise these tools, and parameters have been modified to fit the demand for off-target prediction. CCTop uses Bowtie to find off-target sites by first identifying PAM sites, and matches and mismatches in protospacer sequences are then searched for by bowtie with modified parameters [93]. Up to five mismatches are allowed in the protospacer as more mismatches may prevent DSB induction. CCTop also implements an off-target score for each candidate sgRNA. CHOPCHOP detects off-target sites using Bowtie with parameters -v and -L for searching sgRNA core mit snapgene Crack Key For U [77]. CRISPOR uses BWA to find all potential off-target sites in iterative mode (“-N”), and can find all validated off-targets as well as Cas-OFFinder [8], [36].
In addition to these traditional tools, many other tools and algorithms have been developed for off-target site detection. Cas-OFFinder is one of the most popular tools for searching potential off-target sites, and advantages include no limit to the number of mismatches, PAM types, gRNA mit snapgene Crack Key For U or high running speed with GPUs. Cas-OFFinder can also predict off-target sites with 1 bp deletions or insertions (i.e. DNA/RNA bulges). CasOT is implemented to find Cas9 on-target sites from input sequences, as well as potential off-target sites with up to six mismatches in the seed region (12 nt adjacent to the PAM). This google sketchups can also determine whether off-targets are within a coding exon [108]. Meanwhile, sgRNAcas9 utilises the ultrafast short sequence mapping tool SeqMap [45] to find off-targets, and classifies all sites into three categories to generate a final output of the best candidate gRNAs [110]. Recently, two new alignment-based tools have been developed. Crisflash utilises a tree-based algorithm to rapidly design CRISPR guide RNAs and optimise guide accuracy by incorporating user-supplied variant data [43]. FlashFry rapidly searches off-target sites and provides much useful information (annotation of off-target sites, on/off-target scores, GC content, etc.) for candidate gRNAs [74]. Here we still classified Crisflash and FlashFry as alignment-based methods since they both propose novel algorithms for off-target searching, whereas the scoring approaches they use are borrowed from others [23], [24], [41], [78].
Among these tools, Cas-OFFinder may be the best choice for identifying all potential off-target sites with any Cas nucleases, and FlashFry is also worth a try for its high speed and comprehensive outputs.
3.2. Scoring-based methods
3.2.1. Hypothesis-driven methods
Alignment-based methods are reliable for detection of most potential off-targets, however, mit snapgene Crack Key For U, not all nucleotide positions containing mismatches have the same decisive effect on off-target cleavage. Additionally, alignment-based prediction always outputs redundant off-target sites, many which are false-positives, although users can reduce the number of outputs by restricting MDaemon Email Server Pro Free Download maximum mismatches when exploring off-target cleavage. One study compared experimentally validated off-targets and off-targets predicted by Cas-OFFinder and CCTop, and the results showed that off-targets detected by the prediction tools only covered some of the validated sites, while some off-target sites cannot be predicted solely based on sequence homology [11]. Thus, features that influence the nonspecific binding of CRISPR gRNAs need to be considered to increase the accuracy of off-target detection.
MIT (Hsu-Zhang) score was proposed for off-target evaluation during the early stages of gene editing by CRISPR. Hsu mit snapgene Crack Key For U al. evaluated >700 guide RNA variants and SpCas9-induced indel mutation levels at >100 predicted genomic off-target loci [41]. They evaluated the contributions made by different mismatch positions and numbers in the target 20 bp, and calculated a weight matrix to determine off-target efficiency for each sgRNA. The authors claimed that this score accounts for >50% of the variance in cutting frequency. The MIT score has been integrated into many CRISPR gRNA design tools such as CHOPCHOP and CRISPOR [36], [56].
Cutting frequency determination (CFD) is another popular score for off-target evaluation. It is noticeable that sgRNA can also bind genome loci with non-canonical PAMs such as NAG, NCG and NGA, which may cause off-target cleavage. Doench et al. added PAM features in their scoring metrics [23]. sgRNAs with mismatches and indels in target sequences were also included for examining correlations between sgRNAs and off-targets. CFD score was validated with GUIDE-seq and proved to perform mit snapgene Crack Key For U than MIT score, hence it was adopted by CRISPRscan [78], GuideScan [83] and CRISPOR [36].
The prediction of sgRNA specificity based on the structural features of the Cas9-sgRNA complex proved to be superior to prediction solely based GOM Player 2.3.68.5332 Crack Plus Registration Code 2021 Download sequence features. CRISPRoff [4] and uCRISPR [119] both utilise energetics properties for off-target evaluation. Compared with other scoring methods like MIT and CFD, they both yielded better accuracy in off-target prediction. Nevertheless, neither have not been systematically evaluated by large-scale experiments, and caution should be exercised when using them.
3.2.2, mit snapgene Crack Key For U. Learning-based methods
Sometimes, empirical algorithms cannot effectively evaluate off-targets since they only consider minor features, whereas learning-based methods build complex models using combinations of multiple features, and they may better predict off-targets. In recent years, several new approaches for off-target prediction based on mit snapgene Crack Key For U learning have been developed.
CRISPR Target Assessment (CRISTA) software uses BWA as the off-target search tool and implements multiple features (PAM type, nucleotide composition, GC content, chromatin structure, DNA methylation, RNA secondary structure, etc.) to predict cleavage propensity [1]. CRISTA exhibits better performance than MIT, CCTop and CFD.
The Doench laboratory also developed a linear regression model for prediction of off-target activity called Elevation that takes both sequence and chromatin accessibility features into consideration, as well as observations from others [62]. Elevation predicts individual scores for each off-target site, as well as an aggregate score for gRNAs. Mit snapgene Crack Key For U method performs best among MIT, CFD and CCTop. However, this software can only compute off-target scores for the human exome (GRCh38) and cannot support other organisms on their website.
DeepCRISPR is a state-of-the-art computational platform that unifies sgRNA on-target and off-target site prediction into one framework with deep learning [18]. It identifies all possible sequence and epigenetic features that may affect sgRNA KO efficacy by learning from large data. This method was proved to surpass other available off-target prediction tools [23], [41], [92], [93].
3.3. Benchmark of scoring-based methods
Different scoring methods are based on different characteristics applied by each individual laboratory. In order to compare the accuracy of different scores, Haeussler et al. assessed four hypothesis-driven methods for off-target prediction using the same datasets [36]. CFD score exhibited the best prediction accuracy, whereas CCTop performed the worst. Data imbalance, mit snapgene Crack Key For U, where the number of true observed off-target sites (OTS) identified by off-target detection experiments is much less than that of all potential off-target sites recognized by alignment-based methods, is a common issue in the learning-based methods. To address the problem, a systematic survey was conducted to assess the influence of data imbalance [30]. The authors used bootstrapping sampling and PR-AUC methods to evaluate two well-established models. They emphasized the importance of using symmetric data for model construct and taking unbiased datasets for benchmark. According to current assessment results, we recommend people to use CFD score for off-target prediction. Elevation and DeepCRISPR are suitable for human genome editing, but CRISPRoff and uCRISPR may need further evaluation before use.
4. Tools for CRISPR guide RNA design
A CRISPR/Cas complex typically contains a Cas protein and a sgRNA, both of which determine the cutting activity. However, protein engineering is a complicated and uncertain strategy for most researchers, and optimising gRNAs is more feasible and efficient. A robust gRNA requires maximum on-target efficiency as well as minimum off-target activity. Basing on these two criteria, numerous computational tools have been developed for high-efficiency CRISPR gRNA design. However, each tool possesses distinct features, and user-friendliness is also important. To acquaint users with existing gRNA design tools, we provide an overview of most that are available based on the strength of their features, and this help users to make appropriate selections. Furthermore, we recommend several functional and user-friendly websites for gRNA design (Fig. 2,Table 3).
4.1. A comprehensive overview of CRISPR guide RNA design tools
The CRISPR/Cas system was first adapted for gene editing in 2012, and several tools mit snapgene Crack Key For U developed immediately thereafter, such as Zinc Finger Targeter (ZiFiT) [89], Cas9 guide RNA Design [68] and CRISPR (http://crispr.mit.edu/) [41]. All these platforms have been implemented in the design of gRNAs for model organisms. ZiFiT is also used to design Zinc Finger and TALEN modules, but Cas9 guide RNA Design and CRISPR are now unavailable.
In the following years, various tools for CRISPR gRNA design emerged, due to both urgent demand for these tools, and because people have learned more and more about how the CRISPR system functions and what influences the efficiency and specificity of Cas cleavage. Many of these tools combine multiple scoring methods and provide alternative options for users. Some others have also proposed their own algorithms to rank designed sgRNAs, and this can assist users in gRNA selection. CHOPCHOP [56], [57], [77] provides alternative scores for users such as Rule Set ½ [23], [24], SSC [112], CRISPRscan [78] and deepCpf1 [47]. E-CRISP utilises its own SAE (Specificity, Annotation, Efficacy) score to determine the quality of each sgRNA, while Rule Set 1 [24] and SSC [112] are also included in E-CRISP. CCTop [93] assigns off-target scores for each sequence empirically, while the CRISPRater score [55] is also included to predict the efficiency of sgRNAs. CRISPOR [36] is a versatile platform that ranks gRNAs according to different scores for evaluating potential off-targets in the genome of interest, and for predicting on–target activity.
A large number of CRISPR/Cas-derived RNA-guided Endonucleases (RGENs) have been identified or modified to improve the cutting efficiency and enlarge the editing range. Some tools enable the design of gRNAs for RGENs. For example, Cas-Designer [79] allows users to choose 20 PAM types from different RGENs, while CRISPOR [20] also offers various PAMs from a defined list. To best serve the specialised purposes of different experiments, several web-based tools have been developed. CRISPR-ERA [64] and CHOPCHOP v3 [57] support sgRNA design for the CRISPRa/i system. CRISPETa [85] is mainly used for genome deletion with paired gRNAs, and BE-Designer [42] can be used for designing gRNA for base editing. Recently, three methods have been employed for successively predicting Cas9-editing outcomes: inDelphi [90], FORECasT [5] and Lindel [16]. These approaches can help to identify gRNAs based on forecasting results.
Researchers should choose suitable tools with caution since different tools are in favour of different genomes or cell types. For instance, Yeastriction [72] is specialised for yeast, FlyCRISPR [35] is specific for Drosophila, EuPaGDT [81] is recommended for eukaryotic pathogens, and CRISPR-P [60], [63], CRISPR-PLANT [76], [109] and CRISPR-GE [111] are all implemented for genome editing in plants. Also, user-friendliness is essential for these web-based design tools, mit snapgene Crack Key For U. Based on these considerations, we propose three comprehensive web-based platforms for CRISPR gRNA design.
4.2. Three comprehensive web platforms for CRISPR guide RNA design
4.2.1. CHOPCHOP
CHOPCHOP has a succinct interface with well-rounded functions, >200 genomes are available on the website, and users can provide gene names, genomic coordinates or target sequences as inputs. If a gene is given, users can set specific regions of the gene as targets, such as coding sequences or promoters. Before designing gRNAs, mit snapgene Crack Key For U, two off-target detection methods and seven efficiency scores can be chosen, which aids users in selecting optimal gRNAs for their research.
To satisfy different experimental purposes, mit snapgene Crack Key For U, CRISPR Cas9 nuclease/nickase, Cpf1, CasX, C2C2 and TALEN are all supported by CHOPCHOP, and various modes of DNA targeting are optional such as KO/KI, gene activation/repression, and nanopore enrichment. In activation/repression mode, gRNAs are designed to target the promoter region and its flanking sites in order to bring the activating/repressing domain into close proximity with the transcription start site [17], [54]. Meanwhile, nanopore enrichment mode is implemented to design sgRNAs for long fragment excision within 40 kb. Additionally, the inDelphi model has been integrated into CHOPCHOP for repair profile prediction by Cas9 in five cell types [90].
After clicking the ‘Find Target Sites’ button, a results table is shown, and each candidate gRNA has a rank, genomic information, GC content, self-complementarity score, mismatch number (0–3) and an efficiency score. A graphical view of all gRNAs is also provided in the UCSC Genome Browser, mit snapgene Crack Key For U, which helps users to select optimal gRNAs.
4.2.2. CRISPR RGEN tools
CRISPR RGEN tools, computational tools and libraries for RNA-guided endonucleases, are maintained by the Bae laboratory, and libraries comprise nine useful tools including Cas-OFFinder [8], Cas-Designer [79] and Digenome-Seq [80]. Herein, we mainly discuss the two gRNA design tools Cas-Designer and BE-Designer [42].
Compared with other designer tools, Cas-Designer allows DNA/RNA bulges when performing off-target detection. Additionally, this detection method is more rapid than others due to the Cas-OFFinder algorithm. Genomic sequences or coordinates and fasta file formats are allowed as inputs. Over 350 genomes and 20 PAM types are specified for users, and outputs include target sequences as well as out-of-frame score (calculate by microhomology), mismatch number (0–2) and off-target sites with up to 1 bp bulge. On/off-target sites are redirected to the Ensembl genome browser [26], which is capable of further inspection.
Unlike Cas-Designer, BE-Designer is primarily implemented for base editing. In this tool, four methods of base editing are specified, mit snapgene Crack Key For U, and the editing window region is also adjustable. After the design phase, three outcomes are based on different coding types, and gRNAs, editing window sequences and amino acids are highlighted in a user-friendly manner. This tool is a good choice for base editing.
4.2.3. CRISPOR
Since many tools have been developed for highly efficient CRISPR gRNA design, an ensemble of multiple tools can be useful, and CRISPOR does this well [20], [36]. At present, CRISPOR contains 417 genomes and 19 PAM types, and can meet the needs of most users. CRISPOR takes genome coordinates and sequences <2,000 bp as inputs and provides comprehensive information as outputs. By default, results are shown in two parts; visualisation of PAM sites along the given sequence, which allows for downloading using multiple formats including fasta, GenBank and SnapGene; a table including all information for each predicted gRNA is also generated. In the table, two specificity scores (MIT and CFD) and 10 efficiency scores (Rule Set 2, CRISPRscan, etc.) are calculated [23], [41], [78]. Furthermore, Microhomology and Lindel scores are also provided for outcome prediction [7], [16]. Warnings such as extreme GC content and poly-T values are indicated, the table is downloadable, and candidate off-target sites with up to four mismatches can be visualised and downloaded.
In addition to gRNA design, CRISPOR designs primers for each gRNA as well as off-target sites. These primers are useful when conducting on/off-target validation or in vitro expression experiments. Furthermore, CRISPOR enables filtering of gRNAs with genomic variants based on well-known variant databases or marked input sequences with N characters. Thus, CRISPOR may be the best tool for designing gRNAs.
5. Summary and perspectives
CRISPR/Cas technology has emerged as an advanced strategy for functional genomics, crop breeding and precise medicine. Guide RNAs are indispensable for CRISPR-based editing, and computational approaches provide assistance for efficient gRNA design. Numerous mit snapgene Crack Key For U have been developed for CRISPR gRNA design and evaluation. However, many issues mit snapgene Crack Key For U. For example, experimental datasets used to build models for predicting sgRNA specificity or efficiency are disparate; CRISPRscan score performs worse in mammals than in zebrafish, the genome the predictive model was trained in [36], mit snapgene Crack Key For U. Researchers mit snapgene Crack Key For U therefore know the strengths and weaknesses of each scoring method, and select the optimal tool for their research.
As we become more aware of the mechanisms by which Cas proteins recognise gRNAs, and bind to and cleave target loci, more and more novel features contributing to cutting efficiency and specificity are being identified, including sequence composition. For instance, mit snapgene Crack Key For U, low chromatin accessibility may block access of Cas9, while open chromatin regions are more likely to cause undesired mutations [39], [92]. In practice, specificity is more important than efficiency for application of CRISPR. However, several off-targets remain indecipherable mcafee internet security download Activators Patch current tools, and discovery of novel features may minimise off-target effects.
Too much choice is not always desirable, and an unbiased evaluation can provide guidance. To this end, the Liu laboratory has conducted the benchmarking [30], mit snapgene Crack Key For U, [114], and Haeussler et al. performed a comparison of different predictive scores and integrated most into CRISPOR [36]. None of the tools excel when using heterogeneous data, due to cell-specific or species-specific training models. Therefore, more cell types need to be evaluated. Also, plant and some animal genomes should be analysed thoroughly, since human or mouse have dominated to date. Moreover, it will be beneficial if useful features of multiple tools are mit snapgene Crack Key For U in future software packages.
The major outcomes of Cas9 cleavage is non-random and predictable [102], and several tools have been created for prediction of CRISPR-Cas9 outcomes [5], [16], [90]. Such findings facilitate more accurate gene editing. However, existing tools cannot account for large indels, homozygous sites and the activity of nucleases other than SpCas9, and this should be resolved in future [6].
In summary, the development of computational approaches has revolutionised the design of effective gRNAs, and CRISPR-based gene therapies and customised gene editing may come within reach. It is likely that CRISPR technology will continue to become more powerful in the coming years.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by the National Transgenic Major Project (2019ZX08010003-001-002, 2018ZX08020-003), Jiangsu Province Government Project (BK2018003)/The open funds of the Jiangsu Key Laboratory of Crop Genomics and Molecular Breeding (PL201801) for T.Z. and Y.Z.; the Fund of Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) for T.Z.
References
1. Abadi S., Yan W.X., Amar D., Mayrose I. A machine learning approach for predicting CRISPR-Cas9 cleavage efficiencies and patterns underlying its mechanism of action. PLoS Comput Biol. 2017;13[PMC free article] [PubMed] [Google Scholar]
2. Abudayyeh O.O., Gootenberg J.S., Essletzbichler P., Han S., Joung J., Belanto J.J. RNA targeting with CRISPR-Cas13. Nature. 2017;550:280–284.[PMC free article] [PubMed] [Google Scholar]
3. Adli M. The CRISPR tool kit for genome editing and beyond. Nat Commun. 2018:9.[PMC free article] [PubMed] [Google Scholar]
4. Alkan F., Wenzel A., Anthon C., Havgaard J.H., Gorodkin J. CRISPR-Cas9 off-targeting assessment with nucleic acid duplex energy parameters. Genome Biol. 2018;19[PMC free article] [PubMed] [Google Scholar]
5. Allen F., Crepaldi L., Alsinet C., Strong A.J., Kleshchevnikov V., De Angeli P. Predicting the mutations generated by repair of Cas9-induced double-strand breaks. Nat Biotechnol. 2019;37:64–72.[PMC free article] [PubMed] [Google Scholar]
6. Bae S., Kim J.S. Machine learning finds Cas9-edited genotypes. Nat Biomed Eng. 2018;2:892–893. [PubMed] [Google Scholar]
7. Bae S., Kweon J., Kim H.S., Kim J.S. Microhomology-based choice of Cas9 nuclease target sites. Nat Methods. 2014;11:705–706. [PubMed] [Google Scholar]
8. Bae S., Park J., Kim J.S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30:1473–1475.[PMC free article] [PubMed] [Google Scholar]
9. Boch J., Scholze H., Schornack S., Landgraf A., Hahn S., Kay S. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009;326:1509–1512. [PubMed] [Google Scholar]
10. Bogdanove A.J., Voytas D.F. TAL effectors: customizable proteins for DNA targeting, mit snapgene Crack Key For U. Science. 2011;333:1843–1846. [PubMed] [Google Scholar]
11. Cameron P., Fuller C.K., Donohoue P.D., Jones B.N., Thompson M.S., Carter M.M. Mapping the genomic landscape of CRISPR-Cas9 cleavage. Nat Methods. 2017;14:600–606. [PubMed] [Google Scholar]
12. Cao Q., Ma J., Chen C.H., Xu H., mit snapgene Crack Key For U, Chen Z., Li W, mit snapgene Crack Key For U. CRISPR-FOCUS: a web server for designing focused CRISPR screening experiments. PLoS ONE. 2017;12[PMC free article] [PubMed] [Google Scholar]
13. Mit snapgene Crack Key For U D. Genome engineering with zinc-finger mit snapgene Crack Key For U. Genetics. 2011;188:773–782.[PMC free article] [PubMed] [Google Scholar]
14. Chari R., Mali P., Moosburner M., Church G.M. Unraveling CRISPR-Cas9 genome engineering parameters via a library-on-library approach. Nat Methods. 2015;12:823–826.[PMC free article] [PubMed] [Google Scholar]
15. Chari R., Yeo N.C., Chavez A., Church G.M. sgRNA Scorer 2.0: a species-independent model to predict CRISPR/Cas9 activity. ACS Synth Biol. 2017;6:902–904.[PMC free article] [PubMed] [Google Scholar]
16. Chen Mit snapgene Crack Key For U, McKenna A., Schreiber J., Haeussler M., Yin Y., Agarwal V. Massively parallel profiling and predictive modeling of the outcomes of CRISPR/Cas9-mediated double-strand break repair. Nucleic Acids Res. 2019;47:7989–8003.[PMC free article] [PubMed] [Google Scholar]
17. Cheng A.W., Wang H., Yang H., Shi L., Katz Y., Theunissen T.W. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res. 2013;23:1163–1171.[PMC free article] [PubMed] [Google Scholar]
18. Chuai G.H., Ma H.H., Yan J.F., Chen M., Hong N.F., Xue D.Y. DeepCRISPR: optimized CRISPR guide RNA design by deep learning. Genome Mit snapgene Crack Key For U. 2018;19[PMC free article] [PubMed] [Google Scholar]
19. Chuai G.H., Wang Q.L., Liu Q, mit snapgene Crack Key For U. In silico meets in vivo: towards computational CRISPR-based sgRNA design. Trends Biotechnol. 2017;35:12–21. [PubMed] [Google Scholar]
20. Concordet J.P., Haeussler M. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018;46:W242–W245.[PMC free article] [PubMed] [Google Scholar]
21. Cong L., Ran F.A., Cox D., Lin S.L., Barretto R., Habib N, mit snapgene Crack Key For U. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823.[PMC free article] [PubMed] [Google Scholar]
22. Deltcheva E., Chylinski K., Sharma C.M., Gonzales K., Chao Y.J., Pirzada Z.A. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471:602–607.[PMC free article] [PubMed] [Google Scholar]
23. Doench J.G., Fusi N., Sullender M., Hegde M., Vaimberg E.W., Donovan K.F. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol. 2016;34:184–191.[PMC free article] [PubMed] [Google Scholar]
24. Doench J.G., Hartenian E., Graham D.B., Tothova Z., Hegde M., Smith I. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat Biotechnol. 2014;32:1262–1267.[PMC free article] [PubMed] [Google Scholar]
25. Endo A., Masafumi M., Kaya H., Toki S. Efficient targeted mutagenesis of rice and tobacco genomes using Cpf1 from Francisella novicida. Sci Rep-Uk. 2016:6.[PMC free article] [PubMed] [Google Scholar]
26. Flicek P., Amode M.R., Barrell D., Beal K., Brent S., Chen Y. Ensembl 2011. Nucleic Acids Res. 2011;39:D800–D806.[PMC free article] [PubMed] [Google Scholar]
27. Fu Y.F., Foden J.A., Khayter C., Maeder M.L., Reyon D., Joung J.K. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013;31:822–826.[PMC free article] [PubMed] [Google Scholar]
28. Fu Y.F., Sander J.D., Reyon D., Cascio V.M., Joung J.K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol. 2014;32:279–284.[PMC free article] [PubMed] [Google Scholar]
29. Fusi N., Smith I., Doench J., Listgarten J. In silico predictive modeling of CRISPR/Cas9 guide efficiency. bioRxiv. 2015[Google Scholar]
30. Gao Y., Chuai G., Yu W., Qu S., Liu Q. Data imbalance in CRISPR off-target prediction. Brief Bioinform. 2019:bbz069, mit snapgene Crack Key For U. [PubMed] [Google Scholar]
31. Gasiunas G., Barrangou R., Horvath P., Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria, mit snapgene Crack Key For U. P Natl Acad Sci USA. 2012;109:E2579–E2586.[PMC free article] [PubMed] [Google Scholar]
32. Gaudelli N.M., Komor A.C., Rees H.A., Packer M.S., Badran A.H., Bryson D.I. Programmable base editing of A.T to G.C in genomic DNA without DNA cleavage. Nature. 2017;551:464–471.[PMC free article] [PubMed] [Google Scholar]
33. Gilbert L.A., Horlbeck M.A., Adamson B., Villalta J.E., Chen Y., Whitehead E.H. Genome-scale CRISPR-mediated control of gene repression and activation. Cell. 2014;159:647–661.[PMC free article] [PubMed] [Google Scholar]
34. Graham D.B., Root D.E. Resources for the design of CRISPR gene editing experiments. Genome Biol. 2015;16[PMC free article] [PubMed] [Google Scholar]
35. Gratz S.J., Ukken F.P., Rubinstein C.D., Thiede G., Donohue L.K., Cummings A.M. Highly specific and efficient CRISPR/Cas9-catalyzed homology-directed repair in Drosophila. Genetics. 2014;196:961–971.[PMC free article] [PubMed] [Google Scholar]
36. Haeussler M., Schonig K., Eckert H., Eschstruth A., Mianne J., Renaud J.B. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016;17[PMC free article] [PubMed] [Google Scholar]
37. Harrington L.B., Burstein D., Chen J.S., Paez-Espino D., Ma E., Witte I.P. Programmed DNA destruction by miniature CRISPR-Cas14 enzymes. Science. 2018;362:839–842.[PMC free article] [PubMed] [Google Scholar]
38. Heigwer F., Kerr G., mit snapgene Crack Key For U, Boutros M. E-CRISP: fast CRISPR target site identification. Nat Meth. 2014;11:122–124. [PubMed] [Google Scholar]
39. Horlbeck M.A., Witkowsky L.B., Guglielmi B., Replogle J.M., Gilbert L.A., Villalta J.E. Nucleosomes impede Cas9 access to DNA in vivo and in vitro. Elife. 2016;5[PMC free article] [PubMed] [Google Scholar]
40. Housden B.E., Valvezan A.J., Kelley C., Sopko R., Hu Y., mit snapgene Crack Key For U, Roesel C. Identification of potential drug targets for tuberous sclerosis complex by synthetic screens combining CRISPR-based knockouts with RNAi. Sci Signal. 2015:8.[PMC free article] [PubMed] [Google Scholar]
41. Hsu P.D., Scott D.A., Weinstein J.A., Ran F.A., Konermann S., Agarwala V. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31:827–832.[PMC free article] [PubMed] [Google Scholar]
42. Hwang G.H., Park J., Lim K., Kim S., Yu J., Yu E. Web-based design and analysis tools for CRISPR base editing. BMC Bioinf. 2018;19:542.[PMC free article] [PubMed] [Google Scholar]
43. Jacquin A.L., Odom D.T., Lukk M. Crisflash: open-source software to generate CRISPR guide RNAs against genomes annotated with individual variation. Bioinformatics. 2019;35:3146–3147.[PMC free article] [PubMed] [Google Scholar]
44. Jensen K.T., Floe L., Petersen T.S., Huang J., Xu F., Bolund L. Chromatin accessibility and guide sequence secondary structure affect CRISPR-Cas9 gene editing efficiency. FEBS Lett. 2017;591:1892–1901. [PubMed] [Google Scholar]
45. Jiang H., Wong W.H. SeqMap: mapping massive amount of oligonucleotides to the genome. Bioinformatics. 2008;24:2395–2396.[PMC free article] [PubMed] [Google Scholar]
46. Jinek M., mit snapgene Crack Key For U, Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821.[PMC free article] [PubMed] [Google Scholar]
47. Kim H.K., mit snapgene Crack Key For U, Min S., Song M., Jung S., Choi J.W., Kim Y. Deep learning improves prediction of CRISPR-Cpf1 guide RNA activity. Nat Biotechnol. 2018;36:239–241. [PubMed] [Google Scholar]
48. Kim Y., Cheong S.A., Lee J.G., Lee S.W., Lee M.S., Baek I.J. Generation of knockout mice by Cpf1-mediated gene targeting. Nat Biotechnol. 2016;34:808–810. [PubMed] [Google Scholar]
49. Kleinstiver B.P., Tsai S.Q., Prew M.S., Nguyen N.T., Welch M.M., Lopez J.M. Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells. Nat Biotechnol. 2016;34:869–874.[PMC free article] [PubMed] [Google Scholar]
50. Knott G.J., Doudna J.A. CRISPR-Cas guides the future of genetic engineering. Science. 2018;361:866–869.[PMC free article] [PubMed] [Google Scholar]
51. Koike-Yusa H., Li Y.L., Tan E.P., Velasco-Herrera M.D., Yusa K. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat Biotechnol. 2014;32:267–273. [PubMed] [Google Scholar]
52. Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA mit snapgene Crack Key For U. Nature. 2016;533:420–424.[PMC free article] [PubMed] [Google Scholar]
53. Kuan P.F., Powers S., He S., Li K., Zhao X., Huang B. A systematic evaluation of nucleotide properties for CRISPR sgRNA design. BMC Bioinf. 2017;18[PMC free article] [PubMed] [Google Scholar]
54. La Russa M.F., Qi L.S. The New State of the Art: Cas9 for gene activation and repression. Mol Cell Biol. 2015;35:3800–3809.[PMC free article] [PubMed] [Google Mit snapgene Crack Key For U. Labuhn M., Adams F.F., Ng M., Knoess S., Schambach A., Charpentier E.M. Refined sgRNA efficacy prediction improves large- and small-scale CRISPR-Cas9 applications. Nucleic Acids Res. 2018;46:1375–1385.[PMC free article] [PubMed] [Google Scholar]
56. Labun K., Montague T.G., Gagnon J.A., Thyme S.B., Valen E. CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Res. 2016;44:W272–W276.[PMC free article] [PubMed] [Google Scholar]
57. Labun K., Montague T.G., Krause M., Torres Cleuren Y.N., Tjeldnes H., Valen E. CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 2019;47:W171–W174.[PMC free article] mit snapgene Crack Key For U [Google Scholar]
58. Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359.[PMC free article] [PubMed] [Google Scholar]
59. Langmead B., Trapnell C., Pop M., mit snapgene Crack Key For U, Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10[PMC free article] [PubMed] [Google Scholar]
60. Lei Y., Lu L., Liu H.Y., Li S., Xing F., Chen L.L. CRISPR-P: a web tool for synthetic single-guide RNA design of CRISPR-system in plants. Mol Plant. 2014;7:1494–1496. [PubMed] [Google Scholar]
61. Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760.[PMC free article] [PubMed] [Google Scholar]
62. Listgarten J., Weinstein M., Kleinstiver B.P., Sousa A.A., Joung J.K., Crawford J. Prediction of off-target activities for the end-to-end design of CRISPR guide RNAs. Nat Biomed Eng. 2018;2:38–47.[PMC free article] [PubMed] [Google Scholar]
63. Liu H., Ding Y., Zhou Y., Jin W., Xie K., Chen L.L. CRISPR-P 2.0: an improved CRISPR-Cas9 tool for genome editing in plants, mit snapgene Crack Key For U. Mol Plant. 2017;10:530–532. [PubMed] [Google Scholar]
64. Liu H.L., Wei Z., Dominguez A., Li Y.D., Wang X.W., Qi L.S. CRISPR-ERA: a comprehensive design tool for CRISPR-mediated gene editing, repression and activation. Bioinformatics. 2015;31:3676–3678.[PMC free article] [PubMed] [Google Scholar]
65. Liu X., Homma A., Sayadi J., Yang S., mit snapgene Crack Key For U, Ohashi J., Takumi T. Sequence features associated with the cleavage efficiency of CRISPR/Cas9 system. Sci Rep. 2016;6:19675.[PMC free article] [PubMed] [Google Scholar]
66. Lowder L.G., Zhang D., Baltes N.J., Paul J.W., 3rd, Tang X., Zheng X. A CRISPR/Cas9 toolbox for multiplexed plant genome editing and transcriptional regulation. Plant Physiol. 2015;169:971–985.[PMC free article] [PubMed] [Google Scholar]
67. Lowder L.G., Zhou J., Zhang Y., Malzahn A., Zhong Z., Hsieh T.F. Robust transcriptional activation in plants using multiplexed CRISPR-Act2.0 and mTALE-Act systems. Mol Plant. 2018;11:245–256. [PubMed] [Google Scholar]
68. Ma M., Ye A.Y., Zheng W., Kong L. A guide RNA sequence design platform for the CRISPR/Cas9 system for model organism genomes. Biomed Res Int. 2013;2013[PMC free article] [PubMed] [Google Scholar]
69. Makarova K.S., Grishin N.V., Shabalina S.A., Wolf Y.I., Koonin E.V. A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biology Direct. 2006;1[PMC free article] [PubMed] [Google Scholar]
70. Mali P., Aach J., Stranges P.B., Esvelt K.M., Moosburner Mit snapgene Crack Key For U, Kosuri S. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol. 2013;31:833–838.[PMC free article] [PubMed] [Google Scholar]
71. Malzahn A.A., Tang X., Lee K., Ren Q., Sretenovic S., Zhang Y. Application of CRISPR-Cas12a temperature sensitivity for improved genome editing in rice, maize, and Arabidopsis. BMC Biol. 2019;17:9.[PMC free article] [PubMed] [Google Scholar]
72. Mans R., van Rossum H.M., Wijsman ESET Smart Security 13.2.18.0 Crack + Premium Key (2020), Backx A., Kuijpers N.G., van den Broek M. CRISPR/Cas9: a molecular Swiss army knife for simultaneous introduction of multiple genetic modifications in Saccharomyces cerevisiae. FEMS Yeast Res. 2015:15.[PMC free article] [PubMed] [Google Scholar]
73. Marraffini L.A. CRISPR-Cas immunity in prokaryotes. Nature. 2015;526:55–61. [PubMed] [Google Scholar]
74. McKenna A., Shendure J. FlashFry: a fast and flexible tool for large-scale CRISPR target design. BMC Biol. 2018;16[PMC free article] [PubMed] [Google Scholar]
75. Mendoza B.J., Trinh C.T. Enhanced guide-RNA design and targeting analysis for precise CRISPR genome editing of mit snapgene Crack Key For U and consortia of industrially relevant and non-model organisms. Bioinformatics. 2018;34:16–23. [PubMed] [Google Scholar]
76. Minkenberg B., Zhang J., Xie K., Yang Y. CRISPR-PLANT v2: an online resource for highly specific guide RNA spacers based on improved off-target analysis. Plant Biotechnol J. 2019;17:5–8.[PMC free article] [PubMed] [Google Scholar]
77. Montague T.G., Cruz J.M., Gagnon J.A., Church G.M., Valen E. CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res. 2014;42:W401–W407.[PMC free article] [PubMed] [Google Scholar]
78. Moreno-Mateos M.A., Vejnar C.E., Beaudoin J.D., Fernandez J.P., Mis E.K., Khokha M.K. CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat Meth. 2015;12:982–988.[PMC free article] [PubMed] [Google Scholar]
79. Park J., Bae S., Kim J.S. Cas-Designer: a web-based tool for choice of CRISPR-Cas9 target sites. Bioinformatics. 2015;31:4014–4016. [PubMed] [Google Scholar]
80. Park J., Childs L., Kim D., Hwang G.H., Kim S., Kim S.T. Digenome-seq web tool for profiling CRISPR specificity. Nat Methods. 2017;14:548–549. [PubMed] [Google Scholar]
81. Peng D., Tarleton R. EuPaGDT: a web tool tailored to design CRISPR guide RNAs for eukaryotic pathogens. Microb Genom. 2015;1[PMC free article] [PubMed] [Google Scholar]
82, mit snapgene Crack Key For U. Peng H., Zheng Y., Blumenstein M., Tao D., Li J. CRISPR/Cas9 cleavage efficiency regression through boosting algorithms and Markov sequence profiling. Bioinformatics. 2018;34:3069–3077. [PubMed] [Google Scholar]
83. Perez A.R., mit snapgene Crack Key For U, Pritykin Y., Vidigal J.A., Chhangawala S., Zamparo L., mit snapgene Crack Key For U, Leslie C.S. GuideScan software for improved single and paired CRISPR guide RNA design. Nat Biotechnol, mit snapgene Crack Key For U. 2017;35:347–349.[PMC free article] [PubMed] [Google Scholar]
84. Pickar-Oliver A., Gersbach C.A. The next generation of CRISPR-Cas technologies and applications.
Computational Tools
This list was seeded by the participants of the 2008 workshop on Standards and Specifications in Synthetic Biology.
Please feel free to add information about a computation tool for synthetic biology (CADs, simulators, databases, lab managements systems, automation software, anything) to the list. Place it alphabetically, name the main project contributors, provide a canonical link to the program, and add a paragraph of descriptive text. Please date your entry.
Antimony
http://antimony.sourceforge.net/Lucian Smith, Mit snapgene Crack Key For U Chandran, Herbert Sauro
Antimony is a human-readable and human-writable language for describing biological mit snapgene Crack Key For U. The modules can be connected together by declaring overlapping molecular species between two modules or via the PoPS in/PoPS out interface. The language is similar to the Jarnac language introduced by Herbert Sauro several years ago.
Athena
http://www.washington.edu/staff/deepakc/downloads/InstallAthena.exeDeepak Chandran, Frank Bergmann, Herbert Sauro
Athena is a tool for building, simulating, and analyzing genetic circuits (as well as metabolic/signaling networks, such as SBML files). It provides a visual interface for building biological modules that can be saved and later connected together. The connection can be achieved using either the PoPS interface or by defining overlapping molecular species (similar to the concept of module in CellML and SBML). In addition to simulation, Athena supports a few other useful features: Database of Ecoli regulatory network from RegulonDB, Graphical view of part sequence, Automated derivation of transcription rate equations, Interface to all Systems Biology Workbench programs, Interface with R statistical language, Easy plugin architecture
Note: Athena has been succeeded by Tinkercell
TinkerCell is a highly flexible visual tool. Although still in development, it will have all the features in Athena. In addition, TinkerCell allows a family tree of biological parts to be loaded from a database. TinkerCell comes with a drawing program that allows users to draw their own graphical representations (if they do not like the defaults). C libraries, such as simulations or graph analysis, can easily be incorporated into TinkerCell. New plug-ins can also be added very easily.
BioJade
http://web.mit.edu/jagoler/www/biojade/Jonathan Goler
BioJADE is a design and simulation tool for synthetic biological systems. BioJADE is written in Java, and makes interactive use of BioBrick Repositories. BioJADE enables system designers to specify a system abstractly, tune it, simulate its behavior using a variety of simulators, and finally package the part for use by either the designer or the public.
BioMortar
http://igem.uwaterloo.ca/BioMortarAndre Masella
BioMortar is a lab management system designed specifically to deal with BioBricks. It is also capable of generating cookie-cutter protocols from user-specified templates and tracking the results, including gel images. It is released under the MIT License.
BioStudio
Sarah Richardson, Joel Bader, mit snapgene Crack Key For U, Jef Boeke
BioStudio is both an integrated development environment and a genome version control system, with the ability to modify nucleotide sequences automatically or manually at multiple resolutions. It uses Gbrowse from the GMOD project for its user interface and is currently able to locate and manipulate potential and existing restriction enzyme recognition sites, identify and incorporate unique sequences for PCR identification of wildtype and synthetic sequence, edit existing genome features, and incorporate and annotate user-created genome features. Each version of the genome is encoded in a Gene Feature Format (GFF) file, which is then displayed by the open source annotation viewer GBrowse and stored in a branching version control system, mit snapgene Crack Key For U. Collaboration and transparency is accomplished through the use of a wiki. Each feature in a GFF file will have a corresponding article in the wiki, where registered users can actively discuss its treatment. To ensure that BioStudio actually meets the needs of synthetic biologists, it is under development alongside the design of a synthetic Saccharomyces cerevisiae genome, SC2.0.
BrickIt
http://brickit.wiki.sourceforge.net/Raik Gruenberg and you?
BrickIt aims to create a portable web-based registry that helps synthetic biologists to plan, organize and track their local biobrick samples. The database-backed web server can be downloaded as virtual machine to quickly set up a local registry which coordinates the work within a lab, institute or community. Although the data remain local, the web server itself is an open-source project and new functions or improvements can be easily exchanged between the different local registries. BrickIt thus also offers a platform for the shared development of tools and infrastructure that foster the collaboration within the Synthetic Biology community. BrickIt and everything it relies on are open source and free. BrickIt itself is licensed under the GPL.
Clotho
http://www.clothocad.orgDouglas Densmore, J. Hitman pro 3.8.11 product key Crack Key For U Anderson, Alberto Sangiovanni-Vincentelli
Clotho presents a design environment to manipulate DNA sequence information and store the manipulated data as packaged "parts" back to part repositories. It provides a robust sequence editing environment (highlighting, restriction enzyme library, basic DNA analysis features), a parts management system mit snapgene Crack Key For U browsing, search, and manipulation), and an algorithm manager which allows the introduction of user developed algorithms (currently includes assembly algorithms). The tool is very much in the early stages of development but an alpha release is available. Clotho is part of a larger development of platform-based design tools for synthetic biology. The tool is open source under a BSD license.
Cytostudio
http://moleculamaxima.com/Molecula Maxima
DilutionMagic
http://www.dilutionmagic.org/dilutionMagic
DilutionMagic is a clever dilution calculator which can calculate your serial dilution steps. It can do it for arbitrary concentrations values and arbitrary volumes.
GeneDesign
http://www.genedesign.orgSarah Richardson, Joel Bader, Jef Boeke
GeneDesign is a suite of algorithms that allow users to edit several features of protein coding sequences, including codon usage and restriction enzyme recognition site presence. It will then generate a list of oligos and a road map for the assembly of the sequence by PCR It is mit snapgene Crack Key For U in Perl and is served over the internet; the code is available for local installations. A new, improved version is due before the end of 2008. PMID: 16481661
Gene Designer
https://www.dna20.com/tools/genedesigner.phpDNA2.0
This integrated, stand-alone secure software helps you create DNA constructs on your desktop with unprecedented ease and speed. Available for Mac and PC. For details see Villalobos et al.
GeNetDes
http://soft.synth-bio.org/genetdes.htmlGuillermo Rodrigo, Javier Carrera, Alfonso Jaramillo
GeNetDes is a tool to design transcriptional networks with targeted behavior that could be used to better understand the design principles of genetic circuits. It is a Simulated Annealing optimization algorithm that explores throughout the space of transcription networks to obtain a specific behavior. The software outputs a transcriptional network with all the corresponding kinetic parameters in SBML format. Our tool can also be applied to design networks with multiple external mit snapgene Crack Key For U and output genes. The software, a tutorial manual, parameter sets and examples are freely available in our website. We are currently extending Genetdes to design networks by assembling standardized biological part models. The models contain data obtained from part characterizations. We will evolve such circuits by replacing model parts to reach the imposed design specifications. In addition, we will incorporate the effect of the chassis by including the interaction with the cellular resources.
GenoCAD
http://www.genocad.orgYizhi Cai, Michael Czar, Julie Marchand, Jean Peccoud
GenoCAD is a web-based application guiding users through the design of part-based genetic systems. GenoCAD uses context-free grammars to formalize design strategies for synthetic genetic systems. This approach provides a path to organizing libraries of genetic parts according to their biological functions. It also provides a framework for the systematic design of new genetic constructs consistent with the design principles expressed in the grammar. Using parsing algorithms, mit snapgene Crack Key For U, GenoCAD enables the verification of existing constructs.doi:10.1093/bioinformatics/btm446
Genome Compiler
Genome Compiler is an intuitive all-in-one software platform for life scientists in the genetic engineering, molecular biology and synthetic biology fields, and provides a comprehensive tool for:
- DNA design and visualization
- Data management
- Collaboration platform
- Seamless DNA ordering
Genome Compiler is free for academia users and is available online and in a downloadable version so you can easily access your data on Genome Compiler from anywhere you are. The software supports Windows and Mac. In addition, the software supports common file formats such as: FASTA, Vector NTI, SnapGene, Geneious, Clone Manager, Serial Cloner, Plasma DNA, mit snapgene Crack Key For U, ApE, DNAStar, etc, mit snapgene Crack Key For U.
Metabolic Tinker
http://osslab.lifesci.warwick.ac.uk/Tinker.aspxKent McClymont, Orkun S Soyer
TINKER is a metabolic pathway design/search tool. It compiles mit snapgene Crack Key For U entire set of known reactions and compounds from the latest version of the Rhea database and converts this data into a directed graph. Nodes and edges on this graph correspond to metabolites and reactions, respectively, and thus the entire graph corresponds to the current known metabolic universe. Within this graph, TINKER searches for thermodynamically feasible paths between user defined "source" and "target" compounds and returns the found paths as metabolic pathways (rank-ordered by thermodynamic feasibility)
Operon Calculator
http://salislab.net/softwareDaniel Cetnar, Tian Tian, Iman Farasat, and Howard M. Salis
The Operon Calculator combines 15 biophysical models and design rules to automatically design synthetic operon sequences for maximum tunable control over RNA and protein expression levels. The algorithm also eliminates the presence of several overlapping, undesired genetic elements that will inevitably break the operon's function.
https://salislab.net/software/OperonCalculator_ForwardDesign
ProMoT
http://www.mpi-magdeburg.mpg.de/projects/promot/Katrin Kolczyk, Sebastian Mirschel, Michael Rempel, Mario A. Marchisio
ProMoT is the process modeling tool designed for the convenient setup of synthetic biology models in a modular fashion. Genetic circuits are built just by placing biological parts on a canvas (using drag and drop) and by connecting them through ”wires” that enable flow of signal carriers, as it happens in electrical engineering. ProMoT supports two different modeling approaches -- a quantitative and a qualitative modeling approach. The quantitative approach is based on differential algebraic equations (DAEs) whereas the qualitative approach is a description of the system in the form of logical equations. The final code associated with a circuit can be exported into Matlab or SBML format (Level-1 and Level-2) allowing to run both deterministic and stochastic simulation.
For more detailed information, please refer to the recent papers ProMoT: Modular Modeling for Systems Biology and Computational design of synthetic gene circuits with composable parts or the synthetic biology research page Synthetic Biology at ETH Zurich.
Ribosome Binding Site (RBS) Calculator
http://salislab.net/softwareHoward Salis, Ethan Mirsky, and Christopher Voigt, Nature Biotechnology, v27, 2009
The Ribosome Binding Site (RBS) Calculator is an engineering design method that predicts the translation initiation rate of a protein coding sequence in bacteria. You can use the RBS Calculator to generate synthetic ribosome binding site sequences and to rationally control the production rate of any protein in bacteria from 0.1 to 100,000+ on a proportional scale.
https://salislab.net/software/forward
The RBS Calculator is used by GenScript to synthesize DNA sequences with custom-designed ribosome binding sites.
The RBS Calculator is embedded into Genome Compiler, an intuitive all-in-one software platform for DNA design, which is free for academics to use.
Relevant papers: Translation Rate is Controlled by Coupled Trade-offs between Site Accessibility, Selective RNA unfolding and Sliding at Upstream Standby Sites
A Predictive Biophysical Model of Translational Coupling to Coordinate and Control Protein Expression in Bacterial Operons
Automated Design of Synthetic Ribosome Binding Sites to Control Protein Expression
Riboswitch Calculator
http://salislab.net/softwareBreakthrough Article: Espah Borujeni A., D.M. Mishler, J. Wang, W. Huso, and H.M. Salis. Nucleic Acids Research, mit snapgene Crack Key For U. v44(1). 2016
Riboswitches are shape-changing regulatory RNAs that bind chemicals and regulate gene expression, directly coupling sensing to cellular actuation. The Riboswitch Calculator designs synthetic translation-regulating riboswitches that bind to specific chemicals and activate gene expression. Starting with a known RNA aptamer, the algorithm automatically optimizes the (up to) 30 nucleotide sequences that appear before and after the RNA aptamer to maximize the activation of gene expression. The algorithm combines a statistical thermodynamic model of translation with genetic algorithm optimization. The algorithm's predictions were validated by constructing and characterizing 62 synthetic riboswitches that utilized six different RNA aptamers to sense a diverse range of chemicals (theophylline, tetramethylrosamine, fluoride, dopamine, thyroxine, 2,4-dinitrotoluene). The riboswitches' activation ratios were the mit snapgene Crack Key For U reported to-date for their respective RNA aptamers.
https://salislab.net/software/RiboswitchCalculator_EvaluateMode
Relevant paper: Automated Physics-based Design of Synthetic Riboswitches from Diverse RNA Aptamers
RBS Library Calculator
Iman Farasat, Manish Kushwaha, Jason Collens, Michael Easterbrook, Matthew Guido, and Howard Salis, Molecular Systems Biology, v10(6). 2014
The RBS Library Calculator designs the smallest RBS library sequence that uniformly searches a desired translation rate space, enabling one to efficiently identify optimal protein expression levels in plasmid- or chromosomally-encoded genetic systems. The algorithm has three modes. Search mode allows one to explore the largest possible translation rate space, across a 100,000-fold proportional scale, using the smallest RBS library (16 to 32 variants). Zoom mode allows one to design a minimal RBS library to search a targeted translation rate space between a minimum and maximum. Genome Editing mode designs genomic RBS libraries with a minimal number of consecutive mutations, suitable for designing the mutagenic oligonucleotides needed for MAGE genome mutagenesis. The method has been used to efficiently tune genetic circuits and optimize metabolic pathways.
Relevant paper: Efficient Search, Mapping, and Optimization of Multi-protein Genetic Systems in Diverse Bacteria
https://salislab/software/RBSLibraryCalculatorSearchMode
RoVerGeNe
http://iasi.bu.edu/~batt/rovergene/rovergene.htmGregory Batt, Calin Belta
RoVerGeNe is a software tool for the analysis of dynamical properties of gene networks. Unlike conventional ODE numerical simulation tools, it allows
- to test whether a dynamical property holds for ranges of parameters
- and to find parameter sets for which a given dynamical property hold
The tool is thus useful for robustness analysis and tuning of gene networks. See Bioinformatics paper.
SBW
SBW (Systems Biology Workbench)
SimThyr
http://simthyr.sf.netJohannes W, mit snapgene Crack Key For U. Dietrich
SimThyr is a continuous numeric simulation program for thyroid homeostasis. It is based on a parametrically isomorphic model of the overall system. Applications of this program cover research, including development of hypotheses, and education of students in biology and medicine, nurses and patients.
Relevant papers:
Citation: Dietrich JW (1994-2016) SimThyr 4.0. Fairfax, VA: sourceforge, http://simthyr.sf.net. RRID:SCR_014351.
SynBioSS
http://synbioss.sourceforge.net/Yiannis Kaznessis, Tony Hill, Vassilis Sotiropoulos, Jonathan Tomshine
SynBioSS (Synthetic Biology Software Suite) is a software suite for the quantitative simulation of biochemical networks using hybrid stochastic algorithms. We believe that one shouldn’t need to know how to program (or use command-line) to use sophisticated numerical methods. Through this software, we intend to put the most powerful techniques for simulating chemically reacting networks into the hands of biologists (or any scientist who can put them to good scientific use). SynBioSS can accurately simulate any system modeled as a network of reactions. In order to achieve this result, we wrapped up state-of-the-art algorithms inside a user friendly graphical interface (GUI) that handles input data, runs the simulations and vividly visualizes simulation results, without requiring any programming background from the user. The software is open and runs on any of the three platforms most used by scientists: Windows, Macintosh, and Linux.
Synbiotools
http://synbiotools.orgSwapnil Bhatia (Boston University)
Synbiotools hosts a free and growing software suite for engineering biology including tools for visualization, combinatorial genetic design generation, circuit design, and build automation.
TinkerCell
Engineering platform for building and testing cellular circuits (Deepak Chandran, UW, Seattle)
TinkerCell is an extensible platform for editing and simulating cellular networks. Users can operate the software at different levels including graphical point and click or via an interactive console. While the main interface is visual, programmers may add new features by writing custom programs in C or Python. TinkerCell is designed to incorporate information from database(s), thus the models store information such as rate constants, gene sequences and promoter strengths. Networks can be "modularized" and connected to one another. TinkerCell is cross platform and written in C++. A Python console is provided for interactive control, mit snapgene Crack Key For U.
http://www.tinkercell.com/Home
TinySeq.com
http://tinyseq.comJason Morrison, Mackenzie Cowell
TinySeq is the minimal minimal part storage tool. It assigns a unique url to a given sequence, and stores the sequence's construction format & plasmid. TinySeq supports part composition via the url, so you can get the assembled sequence of two parts simply by asking for something like tinyseq.com/mlc:1+mlc:2. We built tinyseq to reveal what other features besides assigning an accession number (mlc:1) to a sequence would be useful for users at a lab bench who are looking for tools to help them keep their assemblies in order.
PCEnv
http://www.cellml.org/tools/pcenv/Andrew Miller, Justin Marsh, Alan Garny
PCEnv is an environment for creating and simulating arbitrary mathematical models, including mathematical models in the fields of systems and synthetic biology. PCEnv uses CellML as a native format for storing models.
PROTDES
http://soft.synth-bio.org/protdes.htmlMaria Suarez, Pablo Tortosa, Alfonso Jaramillo
Synthetic Biology will benefit from future efforts using first-principles to design biological macromolecules. Ideally, this would mean using the same software and parameters to fold a protein than to design a protein mit snapgene Crack Key For U a given fold (inverse folding problem). Recent work has demonstrated that it is possible to experimentally validate such approaches, by using appropriate physical modelling. We have developed a tool able to incorporate such successful procedures by using a leading molecular dynamics software. Our tool PROTDES is an open-source toolbox for computational protein design using the CHARMM package. This allows the integration of molecular dynamics within the protein design, allowing to extend the physical description more than it has been possible with current software. The procedure automatically finds the suitable mutations optimizing a protein folding free energy. It mutates residue positions to find the best amino acids in an arbitrary protein structure without requiring pairwise approximations. It implements an heuristic optimization algorithm that iteratively searches the best amino acids and their conformations for a an arbitrary set of positions within a structure. The users will be able to create their own procedures for protein design using their own physical protocol, which we exemplify by already incorporating three alternative effective energy functions.Our versatile software will allow synthetic biologists using physical models to use a standard molecular dynamics software for protein design.
j5
http://j5.jbei.orgNathan J. Hillson
Recent advances in Synthetic Biology have yielded standardized and automatable DNA assembly protocols that enable a broad range of biotechnological research and development. Unfortunately, the experimental design required for modern scar-less multipart DNA assembly methods is frequently laborious, time-consuming, mit snapgene Crack Key For U, and error-prone. A web-based software tool, j5, automates the mit snapgene Crack Key For U of scar-less multipart DNA assembly protocols including SLIC, Gibson, CPEC, and Golden Gate. The key innovations of the j5 design process include cost optimization, leveraging DNA synthesis when cost-effective to do so, the enforcement of design specification rules, hierarchical assembly strategies to mitigate likely assembly errors, and the instruction of manual or automated construction of scar-less combinatorial DNA libraries. j5 can be used to build combinatorial libraries and applied to the preparation of linear gene deletion mit snapgene Crack Key For U. These innovations save researchers time and effort, reduce the frequency of user design errors and off-target assembly products, decrease research costs, and enable scar-less multipart and combinatorial DNA construction at scales unfeasible without computer-aided design. The j5 software has been exclusively licensed to TeselaGen Biotechnologies for commercial use and distribution.
DeviceEditor
http://j5.jbei.orgJoanna Chen, Douglas Densmore, Timothy Ham, Zinovii Dmytriv
A web-based bioCAD software tool, DeviceEditor, provides a graphical design environment that mimics the intuitive visual whiteboard design process practiced in biological laboratories. The key innovations of DeviceEditor include visual combinatorial library design, direct integration with scar-less multi-part DNA assembly design automation, mit snapgene Crack Key For U, and a graphical user interface for the creation and modification of design specification rules. DeviceEditor liberates researchers from DNA base-pair manipulation, and enables users to create successful prototypes using standardized, functional, and visual abstractions. Open and documented software interfaces support further integration of DeviceEditor with other bioCAD tools and software platforms. DeviceEditor saves researcher time and institutional resources through correct-by-construction design, the automation of tedious tasks, mit snapgene Crack Key For U, design reuse, and the minimization of DNA assembly costs. The DeviceEditor software has been exclusively licensed to TeselaGen Biotechnologies for commercial use and distribution.
VectorEditor
https://public-registry.jbei.org/static/vesa/VectorEditor.htmlZinovii Dmytriv, Timothy Ham, Hector Plahar, Joanna Chen
Open source, web based cross platform and cross browser DNA sequence editing and analysis tool.
iGEM Software
http://igem.synbioreview.com/Michal Galdzicki
A reference collection of iGEM software projects 2007-2011. This is a compilation of all the software projects large and small created throughout the iGEM competition. On this site you can find the description of the computational tool, a link to the project page and source code.
1
Design Better Procedures
Accurately design and simulate cloning procedures. Test complicated projects, catch errors before they happen, and obtain the right constructs the first time.
Learn More
Cloning is easier when you can see what you are doing. The intuitive interface offers you unparalleled visibility into your work, simplifying often complex tasks.
Learn More
3
Automatically Record Your Work
SnapGene automates documentation, so you don’t have to. See and share every sequence edit and cloning procedure that led to your final plasmid.
Learn More
New and Noteworthy
mit snapgene Crack Key For U Explore SnapGene Academy
Master SnapGene and key concepts in cloning with our new online learning center, SnapGene Academy. Containing over 50 video tutorials taught by scientific experts, SnapGene Academy helps you advance your skills across multiple molecular biology courses.
mit snapgene Crack Key For U More
Discover What’s New in SnapGene 6.1
SnapGene 6.1 provides new functionality for cloning and visualizations. Highlights include simplified primer design when simulating Golden Gate Assembly, a new Secondary Structure view for ssRNA sequences, and support for Dark mode across all operating systems.
Learn More
Coronavirus Resources
Download genomes of common coronaviruses including SARS, MERS and COVID-19, as well as primers and probes for the SARS-CoV-2 genome.
Learn More
SnapGene is mit snapgene Crack Key For U easy to use that my lab adopted it instantly. It saves a lot of time and money as we now do all our cloning in SnapGene first and catch any snags in the strategy before they hold us up.
A. Matouschek
Northwestern University
SnapGene is AMAZING. Designing cloning strategies is incredibly easy and so quick! The In-Fusion simulations have literally saved me days worth of time. The maps look fantastic. I am completely sold!
Richard
Gurdon Institute, University of Cambridge
SnapGene is awesome. It is straightforward enough to start using right away, yet rich with sophisticated features that cover all of my cloning needs. I recommend this software to anyone who clones DNA, mit snapgene Crack Key For U.
D. Strongin
Fred Hutchinson Cancer Center
SnapGene is a most excellent Express Zip For Windows very intuitive software, and I keep discovering mit snapgene Crack Key For U useful features. I have tried MANY software packages, none of them even comes close to SnapGene!
W. Rossoll
Emory University
This application blows everything else past and present out of the water. You all did an AMAZING job making SnapGene powerful and intuitive, yet simple. Excellent work.
Scott
University of North Carolina, Chapel Hill
UCL Software Database
Mathematical
Modelling Software
Statistical
DesktopPC
DesktopPC
DesktopStaff
DesktopPC
DesktopPC
DesktopPC
DesktopPC
Statistical
DesktopPC
Video Mit snapgene Crack Key For U Studio is a screen video capture program
DesktopPC
DesktopPC
DesktopPC
DesktopPC
DesktopPC
DesktopPC
DesktopPC
DesktopPC
DesktopPC
Modelling Software
DesktopPC
DesktopStaff
Staff WTS 2010
DesktopPC
Statistical
DesktopPC
DesktopPC
Mathematical
DesktopPC
Unity
DesktopPC
DesktopPC
Utilities
Mathematical
Programming
Statistical
Utilities
Utilities
DesktopPC
DesktopPC
Utilities
Statistical
Multimedia
Office Suite
Graphic Editing
Office Suite
DesktopPC
Statistical
DesktopPC
Statistical
Unity
DesktopPC
Financial
Programming
Statistical
DesktopPC
DesktopPC
DesktopPC
DesktopPC
DesktopPC
DesktopPC
Staff WTS 2010
DesktopPC
DesktopPC
Utilities
DesktopPC
DesktopPC
DesktopStaff
DesktopPC
Statistical
DesktopPC
Statistical
Utilities
Utilities
Modelling Software
Unity
Modelling Software
Unity
Modelling Software
Programming
Legion
Unity
DesktopPC
Statistical
DesktopPC
Programming
DesktopPC
DesktopPC
DesktopPC
DesktopPC
DesktopPC
DesktopPC
Statistical
Unity
Programming
DesktopPC
DesktopPC
DesktopPC
DesktopPC
DesktopPC
DesktopPC
DesktopPC
DesktopPC
DesktopStaff
DesktopPC
Geographic
Statistical
DesktopPC
DesktopPC
DesktopPC
Mathematical
Programming
Statistical
Capture
Desk Top Publishing
Graphic Editing
Multimedia
Screen Capture
Video Capture
Video Editing
Web Editing
Utilities
Reference Management
DesktopPC
Utilities
Development Tools
Utilities
Diagramming
Utilities
Sound Recording
Video Capture
Video Editing
DesktopPC
DesktopPC
DesktopPC
DesktopPC
DesktopPC
DesktopPC
DesktopPC
Teaching & Learning
DesktopPC
Utilities
Utilities
Document Management
Utilities
DesktopPC
Statistical
Multimedia
Development Tools
Programming
Utilities
DesktopPC
Utilities
DesktopPC
DesktopPC
DesktopPC
DesktopPC
Scanning
Utilities
Modelling Software
Modelling Software
Statistical
Mathematical
Modelling Software
Utilities
Utilities
Utilities
Utilities
Utilities
Reporting Tools
Desk Top Publishing
Graphic Editing
Multimedia
Screen Capture
Video Capture
Video Editing
Web Editing
Reporting Tools
Web Editing
Teaching & Learning
Video Capture
Video Editing
Utilities
Statistical
Utilities
Video Editing
Programming
Utilities
Utilities
Utilities
Video Capture
Video Editing
Utilities
Sound Recording
Utilities
Utilities
Utilities
Utilities
Utilities
geospatial processing & analysis
Modelling Software
geospatial processing & analysis
GIS / Mapping
Modelling Software
geospatial processing & analysis
GIS / Mapping
Modelling Software
Utilities
Project Management
Utilities
-
-
-